
Abstract
Calcitonin gene-related peptide (CGRP), nitric oxide (NO) and histamine are implicated in primary headaches but their role in vascular and nociceptive events in the dura mater is not well described. In an in vitro preparation of the hemisected rat skull, CGRP and histamine release from the cranial dura was measured using enzyme-linked immunoassays. While the NO donator NONOate (10-4 M) was without effect, CGRP (10-5 M) induced considerable histamine release from the rat cranial dura, which was blocked by the CGRP receptor antagonist CGRP8-37 (10-5 M). Conversely, histamine (10-4 M) did not stimulate CGRP release. In vitro recordings from single rat meningeal afferents showed that only one of 12 mechanically identified units but several mechanically insensitive units responded to histamine (up to 10-5 M). Increases in meningeal blood flow after histamine application (10-4 M) to the rat cranial dura remained unchanged during CGRP receptor blockade with CGRP8-37, inhibition of NO synthesis with L-NAME (20 mg/kg i.v.) and H3 receptor blockade with thioperamide (10-4 M). We conclude that histamine produces direct vasodilatation and activates a subset of largely non-mechanically sensitive, non-CGRP containing afferents in the rat meninges. Histamine is released from meningeal mast cells which are stimulated by CGRP. Similar mechanisms may be involved in the pathogenesis of headaches.
Introduction
The endogenous substances calcitonin gene-related peptide (CGRP), histamine and nitric oxide (NO) are all powerful mediators of meningeal and intracerebral vasodilatation and thereby they can potentially produce an increase in meningeal blood flow (1–6). CGRP is released from a population of trigeminal afferents in vivo and from isolated meningeal tissue preparations in vitro in response to appropriate stimuli (7–11). Histamine is released from mast cells, which in the meninges are spatially associated with sensory nerve fibres and blood vessels (12–14). Possible sources of NO in meningeal tissues are endothelial cells, perivascular nerve fibres and immunocompetent cells including mast cells (15, 16). These three mediators of vasodilatation have frequently been implicated in the pathogenesis of migraine and other primary headaches, although the pathological significance of vasodilatation in these diseases remains unclear (17–20). Regarding the classical hypothesis of neurogenic inflammation in the meninges being causal for some forms of headache, a bidirectional communication between sensory nerve fibres and dural mast cells has been suggested (21, 22). Histamine infusion is known to induce headaches preferentially in migraineurs, and this effect has been proposed to be linked to the downstream endogenous formation of NO (20, 23). The vasodilatory effect of histamine has been hypothesized to be partly due to NO production in cranial blood vessels (24).
In previous studies our group has shown that increases in meningeal blood flow caused by different NO donors are attenuated by blockade of CGRP receptors (25, 26). An indirect vasodilatory effect of NO donors in the cranial dura has been confirmed and extended by Akerman et al. (27) using intravital microscopy. They have demonstrated that vasodilatation induced by the NO donor sodium nitroprusside is reduced by inhibiting CGRP release (using the 5-HT1B/D receptor agonist sumatriptan) but not by blockade of H1 and H2 receptors. These two histamine receptors have been shown to be involved in histamine's vasodilatory and blood flow increasing effect in the meninges (28, 29). Furthermore, electrically evoked neurogenic vasodilatation, which is known to depend mainly on CGRP release (3, 6), has been shown by Akerman et al. (27) to be inhibited by high doses of a H1 receptor antagonist, indicating that H1 receptors may also be involved in neurogenic vasodilatation.
To evaluate the role of these multiple interactions in a naturally occurring situation, the endogenous sources of these mediators should be considered. High local concentrations of CGRP and histamine can be expected in the perivascular compartment around CGRP releasing nerve fibres and mast cells. Therefore in the present study we chose local application of CGRP and histamine to examine the influence of these mediators on meningeal blood flow and their effect on the release of one another. We also examined whether functional H3 receptors are involved in histamine-evoked CGRP release as H3 receptors have been shown to inhibit neurotransmitter release from postganglionic sympathetic neurones and neuropeptide release in neurogenic inflammation (30–32). Finally, we used a new preparation of the isolated cranial dura mater to examine whether histamine is able to activate meningeal afferents.
Methods
Preparations ex vivo
Skull preparation
Wistar rats of both sexes were killed by CO2 inhalation and posthumously decapitated. The facial skin and lower jaw were removed, the cranial vault cut mid-sagittally and the cortex and brainstem carefully removed without lesioning the dura mater. The resulting half skull, lined with intact dura, was continuously superfused for at least 30 min with physiological solution (synthetic interstitial fluid, SIF) of the following composition (in mM): Ca2+ 1.54, Cl– 115.3, K+ 3.52, Mg2+ 0.69, Na+ 140, glucose 5.05, buffered to pH 7.4 with carbogen (95% O2/5% CO2).
CGRP and histamine release
Heads were taken from male rats with a body weight of 310–550 g and prepared as described above. The skull halves were mounted in a water bath forming a humid chamber held at a temperature of 37°C. During the experiments the cavities were repetitively filled with SIF that completely covered the supratentorial dura but not the posterior fossa. Five consecutive samples of eluate were collected at intervals of 5 min for CGRP and 10 min for histamine release by evacuating the fluid from the skull without touching the dura. In the first and second sampling period the skull cavity was filled with 300 µl SIF, in the third period with a solution of the test substance in SIF, and in the fourth and fifth sampling period again with SIF. The test substances were histamine (histamine dihydrochloride 10−4 M; Sigma, Taufkirchen, Germany) or a combination of histamine and thioperamide maleate (10−4 M; Tocris, Köln, Germany), Diethylamine NONOate (10−4 M; Calbiochem, Hofheim, Germany) or CGRP (human αCGRP, 10−5 M, Calbiochem). In one series of experiments CGRP8-37 (human CGRP-I 8–37, 10−5 M, Calbiochem) was applied in sampling periods 2–4 and CGRP (10−5 M) was added in the fifth period.
The method for measuring CGRP in the eluates with an enzyme-linked immunoassay (EIA) has been previously published in detail (33). Briefly, the eluates were processed immediately after the experiment using commercial CGRP-EIA buffers and kits (Cayman, distributed by SPIbio, Paris, France). Peptide degradation was prevented by adding EIA-buffer that contains peptidase inhibitors. The antibody contained in the CGRP-EIA kit is directed against human α/β-CGRP but has 100% cross-reactivity to rat and mouse CGRP. CGRP concentrations in the samples were photometrically determined using a microplate reader (Dynatech, Chantilly, VA, USA). The minimum detection level of immunoreactive CGRP (iCGRP) in this assay was about 2 pg/ml.
The histamine measurements were performed with a competitive enzyme-linked immunoassay (IBL, Hamburg, Germany). The eluates were immediately frozen after the experiment. Before processing they were defrosted and incubated in wells together with commercial indicator buffer and acylation reagent for 30 min at room temperature. Then they were again buffered and incubated with enzyme conjugate and histamine antiserum for 3 h at room temperature. After washing with buffer, TMB substrate solution was added to the samples that were incubated for 20 min (room temperature) before the reaction was stopped with TMP stop solution. The optical density was measured at 450 nm using a microplate photometer (Dynatech) and compared with a standard curve obtained with diluted histamine solutions. The detection limit of immunoreactive histamine (iHA) according to the information provided by the manufacturer is about 2.4 ng/ml.
Values of iCGRP or iHA release from subsequent sampling periods were compared using one-way analysis of variance (
Recordings from dural afferents
Heads from rats (180–320 g) of both sexes were prepared as described above. The half skulls lined with intact dura were mounted in a perspex organ bath that was continuously perfused with SIF. The bath temperature was maintained at 35.5 ± 0.3°C by a Peltier element regulated by feedback from a thermocouple in the bath.
Electrophysiological signals from axons innervating the cranial dura were recorded single-ended with a glass micropipette of 10–50 µ
With the recording electrode attached to the spinosus nerve, evidence for impulse activity was sought by washing a small volume of KCl (100 m
Preparations in vivo
Anaesthesia and general preparation
Experimental procedures were in accordance with the regulations and ethical issues for animal care and treatment and received institutional approval by the local district government. Male Wistar rats weighing 280–320 g were anaesthetized by an initial intraperitoneal (i.p.) dose of 120–150 mg/kg thiopentone (Trapanal®, Altana, Konstanz, Germany). Additional doses of thiopentone (25 mg/kg i.p.) were used to hold the anaesthesia at a level in which noxious stimuli (pinching of the earlobes) failed to elicit nociceptive motor reflexes or changes of the systemic arterial pressure. A catheter was inserted into the right femoral vein for the infusion of solutions. Systemic blood pressure was recorded with a pressure transducer via a catheter in the right femoral artery. The body temperature was maintained at 37–37.5°C with a feedback-controlled homoeothermic system (Föhr Medical Instruments, Seeheim, Germany). The animals were tracheotomized and allowed to breathe spontaneously. The room air was enriched with oxygen at the opening of the tracheal tube. This was sufficient to hold ventilation and circulation parameters (inspiration frequency, systemic blood pressure) constant during the whole experiment.
Head surgery and blood flow recordings
The animals were placed in a stereotaxic frame with the head held in a fixed position by ear bars. The eyes were covered with a protecting ointment (Bepanthen®; Roche, Mannheim, Germany). An incision was made along the midline of the scalp, and the left parietal region of the skull was exposed. Using a dental drill and liquid cooling with drops of saline, a cranial window of about 4 × 6 mm was drilled into the parietal bone to expose the dura mater above the middle meningeal artery (MMA). Arterial and venous vessels in the dura mater could clearly be differentiated from cortical arteries. Blood flow was recorded by a laser Doppler system (DRT4, Moor Instruments, Axminster, UK) at a sampling frequency of 1 Hz using needle type probes (tip diameter 0.8 mm) that were orientated with their tips towards branches of the MMA. Recording sites distant from cortical blood vessels were selected along the MMA. In some experiments two probes were used either for parallel or successive measurements at distant MMA branches. The dura in the recording window was protected from drying with pieces of cotton soaked with isotonic saline.
The experiment was started not earlier than 60 min after trepanning the skull to ensure that the basal blood flow was stable. For topical application of substances, the cranial window was filled with 50 µl of solution using an Eppendorf pipette. Histamine (Sigma) dissolved in saline at 10−4 M was applied for 5 min. Following each histamine application the dura was washed and covered with saline to restore the basal flow, which was reached within 10 min of washing. The H3 receptor antagonist thioperamide maleate (10−4 M, Tocris) or the CGRP receptor antagonist CGRP8-37 (human CGRP-I 8–37, 10−4 M, Calbiochem) or both substances dissolved together in saline were locally applied to the exposed dura. Three minutes later the antagonists were carefully removed prior to the test application of histamine (10−4 M, 5 min). The NO synthase inhibitor Nω-nitro-L-arginine methyl ester (L-NAME; Sigma) was slowly injected i.v. over 1 min (20 mg/kg), followed by topical histamine application 8 min later.
The mV readings of the flow monitor were taken as arbitrary units. Control flow values were obtained by measuring the mean flow over a period of 5 min after local application of 50 µl saline (vehicle) to the dura (baseline control). Changes in flow induced by local application of substances were determined as mean flow values (in percentage) within the respective application period (5 or 3 min) relative to the baseline during the control period. Increases in flow after topical application of histamine were calculated as the mean flow within the 5 min following histamine application reduced by the basal flow during the 5-min period before histamine application. Subsequent increases in flow evoked by histamine, before and after application of antagonists, were normalized to the first histamine-evoked flow increase. To compare histamine effects before and after antagonists, one-way
Results
Release of CGRP
CGRP release was measured in 10 hemisected skulls. The mean basal value of iCGRP before histamine stimulation (second sampling period) was 15.2 ± 2.6 pg/ml eluate. There was no correlation of the basal CGRP release with the weight of the animals, as has been seen in previous studies (11, 25). After histamine application (10−4 M) the iCGRP value was 15.3 ± 1.6 pg/ml. When these post-stimulation values were normalized to the basal value within each experiment, it seemed that there was a slight increase (1.24-fold) after histamine but this was not significant. In a further eight experiments histamine (10−4 M) was applied together with the H3 receptor antagonist thioperamide (10−4 M) to unmask a possible H3 receptor effect that may suppress CGRP release. The mean basal iCGRP level was 14.4 ± 1.8 pg/ml and after histamine/thioperamide the iCGRP concentration was 16.9 ± 2.7 pg/ml. These values were not significantly different.
Release of histamine
Histamine release was determined in 10 hemisected rat skulls treated with CGRP. The mean basal level of immunoreactive histamine before application of CGRP was 12.5 ± 0.5 pg/ml eluate without correlation with the weight of the animals. After stimulation with CGRP (10−5 M) the concentration of immunoreactive histamine was 33.1 ± 5.7 pg/ml. This value was significantly different from the values of all other sampling periods (P < 0.005, repeated measures

Histamine (HA) release from the hemisected cranial preparation during successive 10-min sampling periods. The data show mean concentrations of immunoreactive HA (normalized to sample 2) before, during and after application of CGRP. In (b) the CGRP receptor antagonist CGRP8-37 was present during the sampling periods 2–4.
In another set of eight experiments the CGRP receptor antagonist CGRP8-37 (10−5 M) was applied throughout the sampling periods 2–4, and CGRP (10−5 M) was added in the third sampling period. The mean basal level of immunoreactive histamine was 11.0 pg/ml eluate. This value was not changed after application of CGRP8-37 nor was there a significant change after concomitant application of CGRP8-37 together with CGRP (Figure 1b).
Another 10 hemisected skulls were treated with NONOate (10−4 M), a NO donator, which has previously been shown to release CGRP from the dura (25). The basal level of immunoreactive histamine was 13.9 ± 0.7 pg/ml and after application of NONOate the value was 13.3 ± 1.4 pg/ml, i.e. not different from that prior.
Responses of dural afferents to histamine
Activity from 12 mechanically sensitive units in the isolated dura mater was monitored during exposure to progressively increasing concentrations of histamine. The units' mechanical thresholds within their dural receptive fields ranged from 0.4 to 1.25 mN. Most of the receptive fields were located at or close to branches of the middle meningeal artery (Figure 2). Only one of these units was clearly activated by superfusion of histamine at concentrations between 10−8 and 10−5 M (Figure 2c, right column). Following exposure to histamine, the threshold of the mechanically identified units was not significantly altered (paired t-test, n = 12, P < 0.05). However, units for which no mechanical receptive field had previously been determined were on occasion activated by histamine (Figure 2c, left column). Four such units were found. Whether these non-mechanically identified units that responded to histamine were actually mechanically insensitive is not known. In one experiment using electrical stimulation of the dura as an initial searching stimulus, three units (c.v. 2.43, 1.11 and 0.47 m/s, respectively) were simultaneously activated, one of which responded to histamine (10−5 M). None of the three units responded to mechanical stimulation as evidenced by the lack of a marking response during mechanical probing of the dura during repetitive electrical stimulation at 1Hz (see ‘Methods’).

In vitro preparation of the rat cranial meninges showing multiunit spinosus nerve recordings. In each of two separate experiments (left and right columns) a single unit (panel a, spike shape inset) was identified by its response to mechanical stimulation (panel a, thickened black bars) within its dural receptive field (see location 1 for left and 2 for right column). For both experiments the mechanically sensitive units did not respond to the control SIF solution (panel b). However, histamine (panel c, 10−6 M) applied at left most edge of trace activated two units (left column, spike shape insets) that were not previously identified by a mechanical search stimulus and the mechanically sensitive unit for the experiment depicted in the right column. None of the mechanically or histamine sensitive units responded to capsaicin (10−6 M). Bath temperature was maintained by feedback at 36°C throughout each experiment.
Histamine-evoked increases in blood flow
Local application of histamine to the dura mater for 5 min caused reproducible increases in blood flow of 45.3% on average (range 11–93%) without any correlation with the weight of the animals. The magnitude of these responses has previously been shown to depend on the activation of vascular H1 and H2 receptors (4). To examine whether inhibitory H3 receptors are also involved in the compound effect of histamine on the blood flow, the H3 receptor antagonist thioperamide (10−4 M) was pre-applied topically (n = 11 measurements in seven animals). This treatment changed neither the basal blood flow (99.8 ± 0.8% of the baseline control) nor the flow increases evoked by histamine (99.4 ± 2.6% of the control increase).
To examine whether flow increases in response to histamine are partly due to CGRP possibly released from meningeal afferents, the CGRP receptor antagonist CGRP8-37 (10−4 M) was applied prior to histamine (n = 9, five animals). CGRP8-37 itself did not cause significant changes in flow (102.1 ± 4.4% of the baseline control). The response to histamine in the presence of CGRP8-37 was not different to that before CGRP8-37 (96.8 ± 5.8% of the control increase). Furthermore, a combination of CGRP8-37 and thioperamide was pre-applied (n = 8, 5 animals), again without influence on the histamine-evoked flow increases (97.0 ± 5.8% of the control).
In order to examine whether the flow increases evoked by topical histamine application are modulated by the release of NO, the NO synthase inhibitor L-NAME (20 mg/kg i.v.) was administered 8 min prior to histamine (10−4 M) in 11 experiments (eight animals). After L-NAME the basal flow was on average 107.8 ± 5.5% of the baseline control level. The response to histamine after L-NAME was variable but on average not significantly different from the control response before L-NAME (96.4 ± 6.7% of the control increase).
Discussion
The present study was made to examine the interactions of CGRP, histamine and NO with regard to their vascular and neuronal effects in the cranial dura mater. Histamine and CGRP were topically applied to the dura in an attempt to mimic a possible pathophysiological situation of increased release of these substances. The most surprising result was that CGRP stimulates the release of histamine while reciprocally histamine was not able to release CGRP from meningeal afferents, even under blockade of H3 receptors, although histamine excited some dural afferents. The lack of CGRP release is evidenced by the consistency of flow increases induced by histamine in the presence of H3 receptor blockade, CGRP receptor blockade and during inhibition of NO production.
Release of histamine
In the hemisected skull CGRP clearly stimulated histamine release from the dura mater with some variance that may depend on the number of histamine releasing cells. Histamine is most likely released exclusively from degranulated mast cells, as immunohistochemical stainings in the rat dura mater have shown no other histamine-positive structures than mast cells (unpublished data from our group). This is in accordance with previous experiments by Ottosson & Edvinsson (35), who showed a dose-dependent release of histamine from dural mast cells in vitro using CGRP at concentrations of 10-5−10-1 M. A possible effect of substance P on the histamine release from dural mast cells as described by these authors could not be confirmed in the hemisected rat skull (preliminary experiments not published). The histamine releasing effect of CGRP is specific and very likely brought about by CGRP receptor activation as it was blocked by the CGRP receptor antagonist CGRP8-37. The CGRP receptors mediating histamine release are probably localized on mast cells, as indicated by new immunocytochemical data with an antibody directed against the CRLR part of the CGRP (unpublished results from our group). The CGRP-stimulated histamine release was rather variable, evidenced by the large standard error of the mean (see Figure 1a). In some samples there was nearly no stimulated histamine release, in others there was a four- to five-fold increase compared with the basal level. This variance, which may indicate different numbers of mast cells in the dura or different states of sensitivity to degranulating stimuli, is also seen with other mast cell degranulators like codeine (results from our group to be published).
Mast cells in the rat dura are located close to sensory nerve fibres (21). Mast cell degranulation has been suggested to be one important element of neurogenic inflammation when meningeal afferents are antidromically activated (14). In this model of neurogenic inflammation signs of degranulation have morphologically been demonstrated although histamine levels have not been measured.
Responses to histamine
Histamine may be involved in causing plasma leakage from post-capillary venules (36, 37) although inhibition of mast cell degranulation alone does not prevent neurogenic inflammation in the dura (38). The main vascular effect of histamine, namely vasodilatation causing increased perfusion of the dura mater, is controlled by H1 and H2 receptors of arterial vessels (4, 29). While H2 receptors on smooth muscle cells mediate vasodilatation, activation of smooth muscle H1 receptors causes vasoconstriction. The vasodilatory effect of H2 receptors dominates when histamine is locally applied to the dura, i.e. when its concentration is high in the perivascular compartment where the mast cells are located. In contrast to the perivascular histamine, high histamine levels in the blood stream activate endothelial H1 receptors that promote endothelial NO synthase activity to produce NO, which diffuses to the smooth muscle cells and causes vasorelaxation (39). This is why blockade of smooth muscle H1 receptors after topical application of H1 receptor antagonists onto the dura increased the blood flow stimulating effect of topical histamine (4), while blockade of endothelial H1 receptors or inhibition of the NO synthase reduced the vasodilatory effect of histamine when it was injected i.v. (29). It is therefore not surprising that in our experiments the blood flow increasing effect of topically administered histamine acting directly on the smooth muscle could not significantly be altered by NO synthase inhibition in the endothelium. The considerable variation in these experiments may nevertheless indicate that histamine could partly have diffused into the blood vessels. Alternatively, we cannot exclude interactions of histamine with other cellular sources of NO such as mast cells and dural nerve fibres, probably of trigeminal and parasympathetic origin (40, 41), in which NO synthase immunoreactivity has also been localized (16). Several functional studies suggest that neurogenic vasodilatation and blood flow increases in the dura induced by electrical stimulation depend in part on the production of NO (1, 29, 42, 43).
NO donors have been shown at high concentrations to release CGRP from the rat dura mater (25) and to either sensitize or desensitize meningeal afferents to mechanical stimuli (44). However, CGRP containing meningeal afferents are probably not those activated by histamine superfusion of the isolated dura in our experiments, because histamine, in contrast to NO, was not able to release CGRP. A major proportion of meningeal afferents cannot be expected to release CGRP, as CGRP immunoreactivity has been found in only one-third of trigeminal ganglion cells projecting to intracranial vessels (45). Therefore we postulate at least two types of meningeal afferents, one type that may be sensitive to NO and can release CGRP and another type that is sensitive to histamine but does not release CGRP. These two morpho-functional types of primary afferents may also exist in the spinal system, as about 15% of lumbar dorsal root ganglion cells in the guinea that express H1 receptor mRNA were not immunoreactive to the neuropeptides CGRP and substance P (46).
The NO donor NONOate, although it releases CGRP from the dura (25), did not measurably stimulate histamine release, which was stimulated by direct application of high doses of CGRP in our experiments. This is probably a matter of concentration, as the released CGRP is rapidly diluted by the superfused physiological solution, in which it was measured at a final concentration in the order of about 10−11 M. The local concentration of released CGRP as well as histamine in the perivascular space of the intact dura can be estimated to be much higher but further studies are needed to clarify these issues.
The following scenario summarizes the hypothetical cascade of nociceptive events in the dura mater (Figure 3). Endothelial NO at low concentrations constitutively contributes to vascular relaxation. At high concentrations it may reach perivascular afferents and facilitate CGRP release. Mast cells in the vicinity of the afferent terminals are activated by CGRP and release histamine, which in turn activates histamine-sensitive afferents. All three chemical mediators contribute to vasodilatation, and this may further facilitate endothelial (and possibly extravascular) NO formation. The consequence is a vicious circle that may finally lead to a significant activation of trigeminovascular afferents.
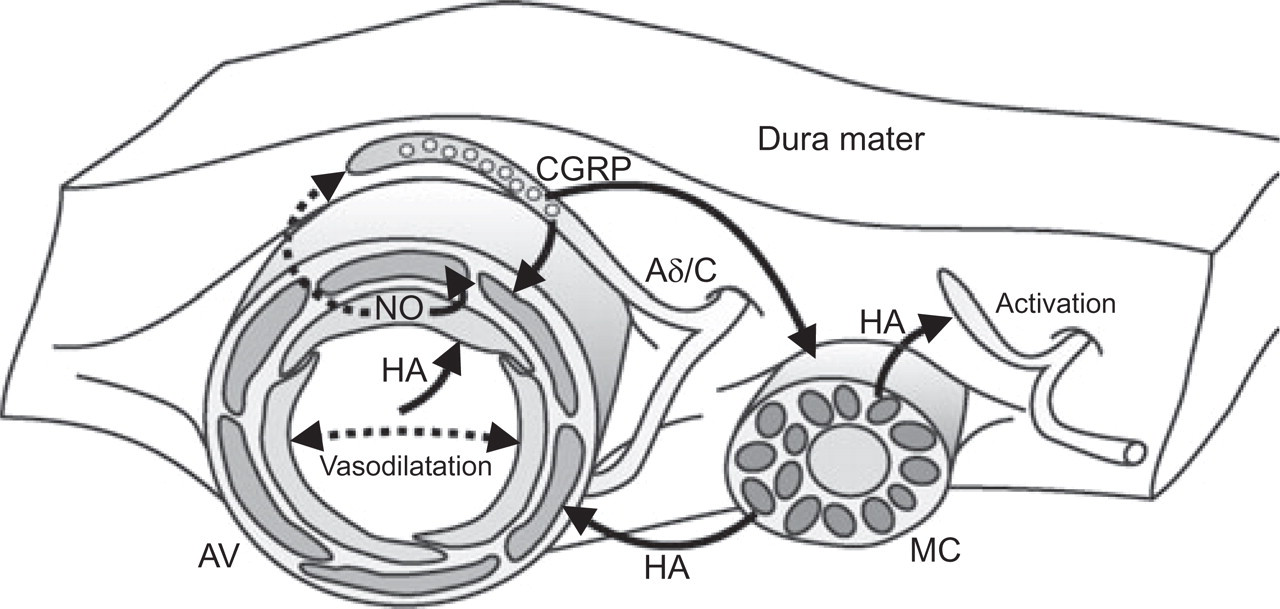
Proposed cascade of mediators and their effects in the dura mater. Terminals of Aδ and C afferents approaching arterial vessels (AV) release CGRP upon activation, probably facilitated by nitric oxide (NO) from vascular endothelium and other sources. CGRP releases histamine (HA) from dural mast cells (MC), and is in turn able to activate a population of afferent fibres. HA may release NO from endothelial cells if it is present in the arterial lumen. All mediators are powerful arterial vasodilators.
Relevance for the pathogenesis and treatment of headaches
NO, histamine and CGRP have been shown to cause headaches preferentially in migraineurs and other patients suffering from primary headaches (23, 47, 48). Originally it had been proposed that histamine might cause these headaches through H1 receptor mediated release of NO from intracranial blood vessels, because blockade of H1 receptors prevented histamine-evoked but not NO-induced headaches (49, 50). However, histamine infusion has more recently been found to dilate intracerebral arteries and to cause headache in migraineurs despite inhibition of endogenous NO synthesis (20). In the cascade of nociceptive events depicted in Figure 3 histamine has an intermediate position in that it is released by CGRP on the one hand and activates meningeal afferents (without releasing CGRP) on the other, probably through activation of H1 receptors. Histamine H1 receptor inhibition could therefore be assumed to be helpful in the treatment of headaches but to date this has not been shown to be beneficial. Instead, specific activation of inhibitory H3 receptors has recently been suggested as a new option for migraine prophylaxis (51). Inhibition of CGRP receptors by the non-peptide CGRP receptor antagonist BIBN4096BS (52) has also recently been shown to be promising in migraine therapy (53). This indicates that CGRP, which is able to release histamine from meningeal mast cells and itself acts as a very potent vasodilator of intracranial arteries (54), has a prominent position in the nociceptive cascade of primary headaches. It should be considered, however, that apart from the meninges there may be several other targets of NO, histamine and CGRP, including the trigeminal ganglion and trigeminal brainstem nuclei, which may exhibit receptor sites for these mediators and are likely to be involved in nociceptive mechanisms underlying the generation of headaches.
Acknowledgements
We thank I Izydorczyk, A Kuhn, J Schramm, M Schulte and B Vogler for their excellent technical assistance. This study was supported by the DFG (SFB 353), the Wilhelm-Sander-Stiftung and the BMBF (German Headache Consortium). M Dux and R Carr were fellows of the Alexander von Humboldt-Stiftung.