
Abstract

A 49-year-old man in otherwise good health presented for two types of headache, both present for 3 years and both confined to the right side. His clinical and neurological examinations were unremarkable. The first headache type was characterized by orbital and fronto-parietal episodes of excruciating pain lasting about 1 h, not accompanied by autonomic manifestations or agitation. There were one to three of these attacks per day, mainly in the morning and at night; they responded poorly to triptans.
The second headache type was characterized by severe burning orbital pain episodes, seven to eight per day but never at night, lasting on average 1 min (range 1–5 min) and again not associated with autonomic phenomena. There were no triggering factors.
Cerebral computed tomography (CT), performed elsewhere at onset 3 years previously, had revealed nothing abnormal; the patient had been diagnosed with ‘cluster-like headache’ and was prescribed verapamil (80 mg three times a day for 3 months) without benefit. We concurred with the ‘cluster-like’ diagnosis and increased verapamil to 120 mg three times a day, with regression of the cluster-like attacks but persistence of the brief duration headaches.
We performed cerebral magnetic resonance imaging (MRI) with contrast which revealed an intrasellar lesion expanding into the inferior and lateral right sellar regions, suggesting pituitary adenoma (Fig. 1). Serum prolactin was 3500 ng/ml (normal range 0.33–27.3 ng/ml), but levels of other hormones were normal. Visual field, optic fundi and ocular mobility were normal. History taking did not reveal alterations in libido. Treatment with the dopaminergic agonist cabergoline normalized serum prolactin but had no effect on the headaches or the size of the lesion. Since the lesion surrounded the internal right carotid and was impinging on the optic chiasma, transnasal adenomectomy was performed in September 2002. Examination of the specimen confirmed adenoma and showed numerous prolactin (PRL)-positive cells; immunostaining for other hormones, in particular growth hormone (GH) and adrenocorticotrophic hormone (ACTH), was negative. There were no sequelae to the operation, immediately after which both headaches disappeared. Seventeen months later the patient remains pain free.
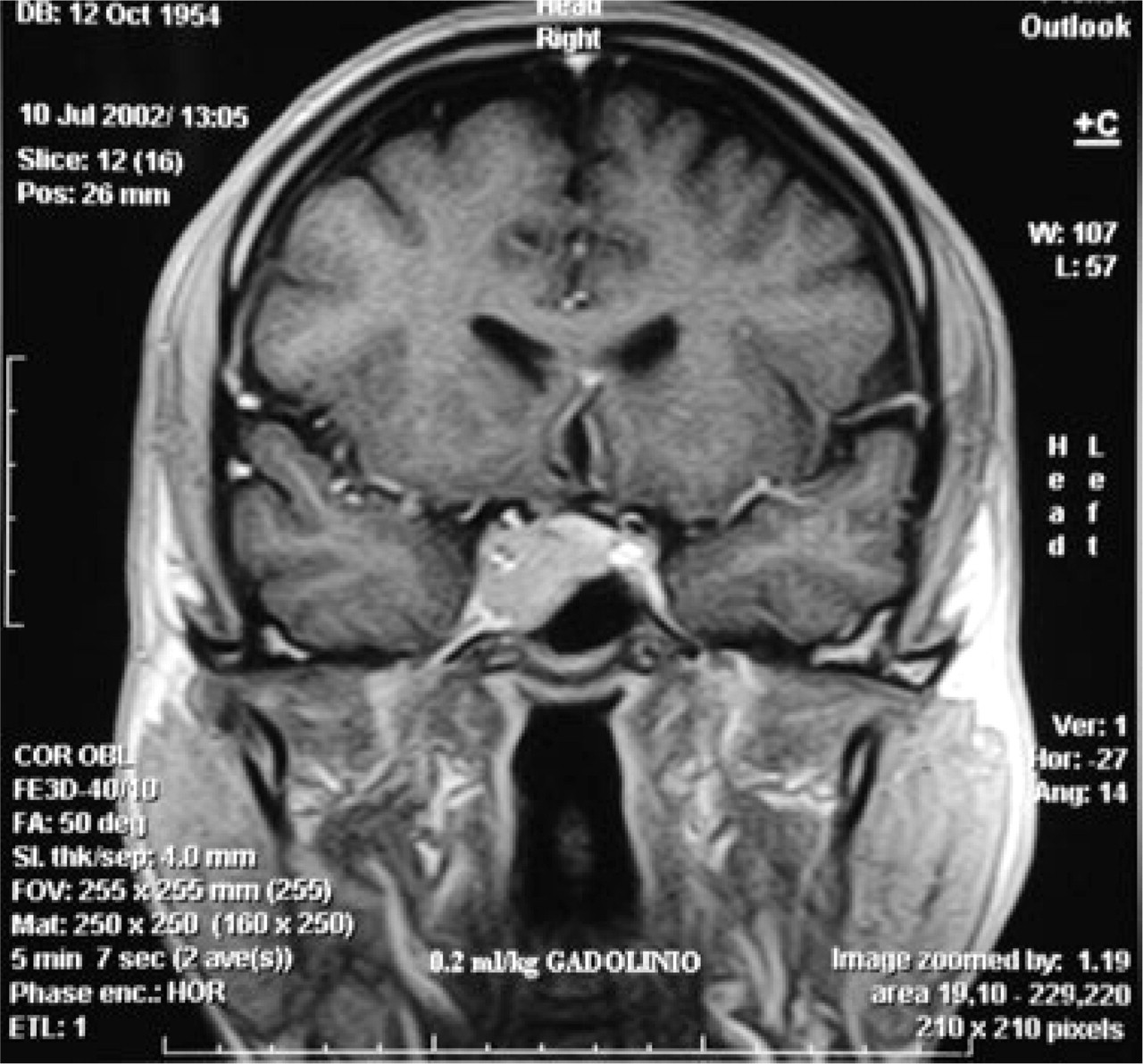
T1-weighted brain MRI with contrast showing intracellular lesion expanding into the inferior and lateral right sellar regions.
We diagnosed the headache with episodes lasting about 1 h as cluster-like disorder not fulfilling the criteria, in view of the absence of autonomic phenomena [chronic cluster headache (CH) without autonomic features]. The second, brief-duration headache, was trigeminal neuralgia, which although fulfilling the International Headache Society criteria (1), was highly unusual for the total absence of triggering factors, longer than normal duration (more than ‘a few seconds’ (1)) and low attack frequency. SUNCT was excluded for absence of autonomic phenomena; idiopathic stabbing headache was excluded as the episodes lasted longer than a few seconds.
The CH–trigeminal neuralgia association, known as cluster–tic, is a rare condition in which CH and trigeminal neuralgia are present on the same side. In four patients with this condition operated on to free the trigeminal nerve from impinging blood vessels, only the neuralgia improved (2). In our patient adenomectomy resulted in resolution of both headaches. To our knowledge, this is the first reported case of successful surgical treatment of both pain components of cluster–tic.
Trigeminal neuralgia is highly unusual in pituitary adenomas. In a series of 2000 trigeminal neuralgias, the cause was pituitary adenoma in only one case (3) in which adenoma removal resulted in neuralgia disappearance (4). Several cases of CH associated with pituitary adenoma have been reported. In such cases adenomectomy completely resolved the pain episodes (5, 6).
In our patient, both headaches are reasonably attributable to the adenoma per se, since: the patient had never suffered from headache before onset of the present condition; both headaches appeared at the same time and were of high frequency; both disappeared immediately after surgery and did not recur in the ensuing 17 months; furthermore, treatment with cabergoline normalized serum prolactin but had no effect on the headaches. This latter finding indicates that abnormal levels of this hormone did not play a role in generating either of the two headache forms. The lesion occupied the right cavernous sinus and surrounded the ipsilateral internal carotid. We propose that traction on sensitive trigeminal fibres in the sinus (the ophthalmic branch of the trigeminal and the dura) or in the internal carotid wall gave rise to both painful conditions.
Primary CH and trigeminal neuralgia have different pathophysiologies (7). In the present symptomatic case it seems that stimulation of the trigeminal fibres, peripherally, gave rise to both headaches. This contrasts with the situation in primary CH, where the trigeminal seems to be activated by the hypothalamus (antidromic mechanism) (7), although some still consider that primary CH may be due to cavernous sinus abnormalities (8). CH attacks are well known in patients with pituitary adenomas and the pain attacks typically disappear after adenoma removal (4–6). This observation suggests that symptomatic CH may be triggered by peripheral trigeminal structures.
The characteristics of the headaches in our patient (absence of autonomic symptoms, late onset, chronic from the beginning) should have led to the suspicion of an underlying rather than essential cause at the outset, and in fact CT was performed. However, only cerebral MRI was able to demonstrate the lesion. This suggests that in the presence of atypical CH-like headache, and unusual trigeminal neuralgia, the patient should be closely followed and that MRI rather then CT should be the neuroimaging modality of choice.