
Abstract
Zebrafish is an ideal system to study the effect(s) of chemical, genetic, and environmental perturbations on development due to their high fecundity and fast growth. Recently, single-cell sequencing has emerged as a powerful tool to measure the effect of these perturbations at a whole-embryo scale. These types of experiments rely on the ability to isolate nuclei from a large number of individually barcoded zebrafish embryos in parallel. Here, we report a method for efficiently isolating high-quality nuclei from zebrafish embryos in a 96-well plate format by bead homogenization in a lysis buffer. Through head-to-head single-cell combinatorial indexing RNAseq experiments, we demonstrate that this method represents a substantial improvement over enzymatic dissociation and that it is compatible with a wide range of developmental stages.
Zebrafish is a useful organism for the study of gene regulation in development. Recently, multiple large-scale single-cell atlases of zebrafish embryonic development have been published, allowing for the efficient annotation and analysis of new single-cell datasets.1,2 These atlases, coupled with new techniques for barcoding and multiplexing individual embryos can be leveraged to perform high-throughput reverse genetics experiments at single-cell resolution. 2 For example, studies have leveraged these techniques to conduct time course experiments on the effects of chemical, environmental, and genetic perturbations on zebrafish development at scale.2,3 A critical first step in multiplexed single-cell experiments is the efficient isolation of high-quality nuclei from uniquely barcoded embryos in a plate format. Previous studies used an enzymatic and mechanical dissociation approach that requires variable amounts of pipetting at elevated temperatures.2,4 As an alternative, we developed a new multiplexed mechanical dissociation method utilizing bead homogenization in lysis buffer 5 (Fig. 1A, Supplementary Data S1). Compared to enzymatic dissociation, our method is faster, less prone to experimental variability, better suited for a wider range of stages, and produces a greater number of nuclei that perform better in downstream sequencing protocols.
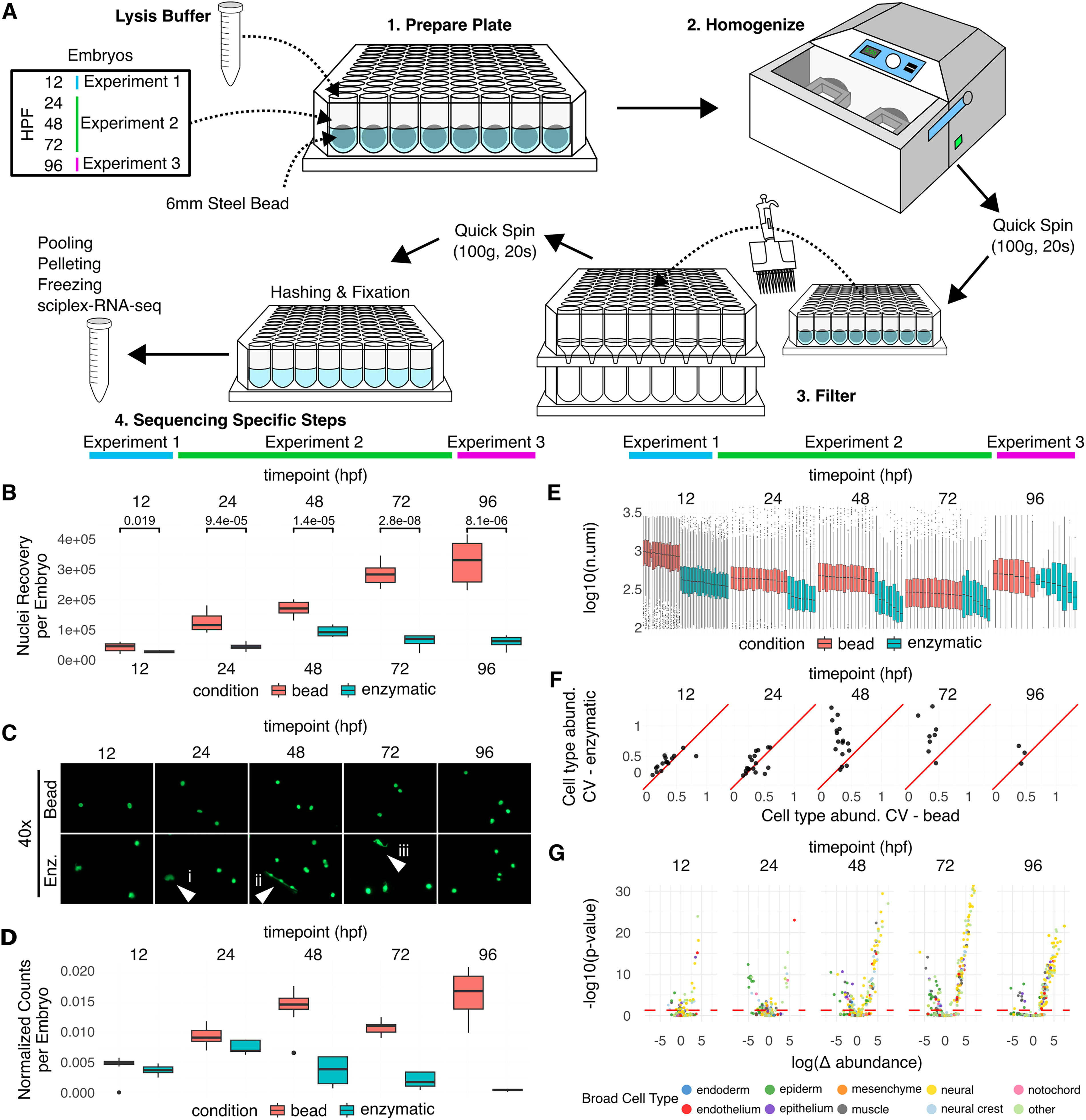
In order to assess the performance of bead homogenization against enzymatic dissociation, we isolated nuclei using each method at 12 hours post fertilization (hpf) (experiment 1), 24 hpf, 48 hpf, and 72 hpf (experiment 2), and 96 hpf (experiment 3) (Fig. 1A, Supplementary Data S1, Supplementary Data S4). We found that prior to sequencing, bead homogenization recovered more nuclei at each time point sampled, with roughly five times more nuclei recovered at 96 hpf (5.53-fold change, p = 8.1e-0.6, n = 8 per condition) (Fig. 1B). Enzymatic dissociation requires variable amounts of time pipetting at 37°C, with longer amounts of time required in older embryos. Longer exposure to the enzymes may explain why the number of nuclei recovered does not increase after 48 hpf when using enzymatic dissociation. Nuclei recovery using bead homogenization; however, scales with developmental stage, suggesting that bead homogenization may be more effective at isolating nuclei from embryos at later stages than those tested in this study (Fig. 1B). To determine the effects of each method on nuclei morphology, we took images of nuclei collected at each time point. We observed that enzymatic dissociation results in small pieces of tissue that are not completely dissociated, nuclei that have tissue or extra cellular matrix still attached, and damaged nuclei that have presumably undergone too much enzymatic dissociation or experienced too much heat stress. (Fig. 1C). In summary, bead homogenization recovers more nuclei per embryo and creates less debris.
On the nuclei isolated at each time point, we then conducted three single-cell combinatorial indexing RNAseq 6 (sciPlex-RNA-seq) experiments comparing our new bead homogenization method to the enzymatic method (Fig. 1A). In these experiments, we used a similar embryo labeling strategy as Saunders et al. 2023, 2 allowing us to pool embryos prior to performing sciPlex-RNAseq, following the Martin et al. 2023 6 protocol with slight adaptations for working with zebrafish nuclei. Even after normalizing the number of nuclei used as input to sequencing, we detected substantially more nuclei isolated by bead homogenization, especially for older embryos (Fig. 1D). From these nuclei, we also recovered more unique molecular identifiers (UMIs) per nucleus, indicating higher RNA quality (Fig. 1E). Due to the multiplexing of individual embryos, we were also able to assess the embryo-to-embryo variability in cell type abundance within each experiment and show that bead homogenization resulted in less within-experiment variability (Fig. 1F). Utilizing software for computing changes in cell type abundance (https://cole-trapnell-lab.github.io/hooke/), we then investigated if there was any bias in cell type recovery between bead and enzymatic dissociation. We observed that nuclei isolated by bead homogenization were broadly enriched for neurons, while nuclei isolated by enzymatic digestion were broadly enriched for epithelial cell types. These biases became more pronounced after 24 hpf, coinciding with the rapid increase of neurogenesis. 7 However, due to a lack of ground truth data, we are unable to determine which method more accurately reflects the true cell type proportions. We caution users to take these biases into account when isolating nuclei for answering specific questions. These results demonstrate that nuclei isolated by bead homogenization are more robust to the library prep, and cellular abundance is more consistent from embryo to embryo, but that there exists a cell type bias when compared to enzymatic digestion methods.
While this protocol serves as an outline for nuclear isolation by bead homogenization for sciPlex-RNA-seq in zebrafish, we anticipate that these methods can be readily adapted to additional embryo species (or embryo models), as well as to accommodate a number of other applications. For example, in preliminary tests, we isolated nuclei using other lysis buffers. We have not tested nuclei isolated by bead homogenization with microfluidic-based single-cell RNAseq platforms, and additional centrifugation or filtration may be required because of the fluidics sensitivity to debris in the sample. We also suspect that this protocol will be broadly applicable to embryos from other organisms, and there is precedent for bead homogenization’s use in mammals in a nonmultiplexed format. 8 To facilitate optimization for new applications, we provide a troubleshooting guide of common problems we encountered and how to solve them (Supplementary Data S2), as well as an online version of the protocol to allow for updated optimizations to be rapidly shared. 5 One barrier to the adoption of bead homogenization may be the cost of the homogenizer (∼$10,000). However, in proof-of-concept testing, we were able to use the less expensive QIAGEN TissueLyser LT Bead Mill to dissociate embryos from tubes. This resulted in similar nuclei recovery and successful sequencing (data not shown), but it is not as amenable to multiplexed applications. Overall, the results of our experiments demonstrate that bead homogenization produces a greater number of higher-quality nuclei per embryo, a greater number of UMIs per nuclei, and less variability between replicates, improving the efficiency, quality, and reproducibility of multiplexed single-nuclei sequencing experiments on zebrafish embryos.
Footnotes
Acknowledgments
The authors thank Dr. Lauren Saunders, Dr. Sanjay Srivatsan, and Beth Martin for helpful discussions, as we were developing this method.
Authors’ Contributions
C.R.: Conceptualization, methodology, validation, formal analysis, investigation, resources, writing—original draft, writing—review and editing, visualization; H.L.: Conceptualization, methodology, validation, formal analysis, investigation, resources, writing—original draft, writing—review and editing, visualization; A.T.: Methodology, validation, investigation, writing—review and editing; R.D.: Conceptualization; A.M.: Investigation, writing—review and editing; J.S.: Supervision, funding acquisition; D.K.: Resources, writing—review and editing, supervision; C.T.: Writing—review and editing, supervision, funding acquisition.
Disclosures Statement
C.T. is a scientific advisory board member, consultant and/or co-founder of Algen Biotechnologies, Altius Therapeutics and Scale Biosciences. J.S. is a scientific advisory board member, consultant and/or co-founder of Cajal Neuroscience, Guardant Health, Maze Therapeutics, Camp4 Therapeutics, Phase Genomics, Adaptive Biotechnologies, Scale Biosciences, Sixth Street Capital, Prime Medicine, Somite Therapeutics and Pacific Biosciences. One or more embodiments of one or more patents and patent applications filed by the University of Washington may encompass methods, reagents, and the data disclosed in this article. Inventors on these patents include C.T.
Funding Information
This work was supported by the National Institutes of Health (RM1HG010461 to C.T., R01HG012761 to C.T. and D.K., and R01HG010632 to C.T. and J.S.) the Paul G. Allen Frontiers Group (Allen Discovery Center for Cell Lineage Tracing to C.T. and J.S.), and the Seattle Hub for Synthetic Biology, a collaboration between the Allen Institute, the Chan Zuckerberg Initiative (award number CZIF2023-008738), and the University of Washington.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.