
Abstract
Abstract
Zebrafish embryos have been widely used to study the genes and processes needed for normal vertebrate heart development. We recently observed that exposure to 2,3,7,8-tetra-chlorodibenzo-p-dioxin (TCDD) or retinoic acid (RA) produces very similar signs of heart failure in developing zebrafish via divergent molecular pathways. The fact that diverse stressors and mutations cause severe pericardial edema and circulatory collapse in developing zebrafish has been largely unexplored. We hypothesized that unrelated chemicals can trigger a common pathological response leading to the same end-stage heart failure. To test this hypothesis, we compared the effects of TCDD, RA, carbaryl, valproic acid, and morpholino oligonucleotide (MO) knockdown of TBX5 on the developing heart in zebrafish embryos. These model stressors have all been previously reported to affect zebrafish heart development, and elicited very similar signs of embryonic heart failure. Microarray analysis showed that one cluster of 92 transcripts affected by these different treatments was significantly downregulated by all treatments. This gene cluster is composed of transcripts required for chromosome assembly, DNA replication, and cell cycle progression. We refer to this cluster as the cell cycle gene cluster (CCGC). Immunohistochemistry revealed that downregulation of the CCGC precedes a halt in cardiomyocyte proliferation in the hearts of zebrafish exposed to any of the treatments. Previous work has shown that the initial response to TCDD is a decrease in cardiac output. Since this precedes the signs of edema, heart failure, and fall in CCGC expression, we postulated that any factor that decreases cardiac output will produce the same syndrome of heart failure responses. To test this, we used MO knockdown of cardiac troponin T2 (TNNT2) to specifically block contractility. The TNNT2-MO produced exactly the same signs of cardiotoxicity as the other treatments, including downregulation of the signature CCGC. Our results indicate that agents altering cardiac output can have amplified consequences during specific periods in development.
Introduction
Zebrafish are a model for studying cardiovascular disease
Tetra-chlorodibenzo-p-dioxin and retinoic acid cause cardiotoxicity
Previous findings in our laboratory showed that, when zebrafish embryos were exposed to either retinoic acid (RA) or 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) they exhibited a similar set of cardiotoxic responses. This included the formation of a small and elongated heart, circulatory failure, and severe pericardial edema.3–5 RA and TCDD exert their effects on zebrafish embryos through very divergent mechanisms, but both cause a similar embryonic cardiac end-stage phenotype.
TCDD activation of the aryl hydrocarbon receptor (AHR) causes translocation of cytoplasmic AHR into the nucleus where it forms a heterodimer with a related protein called ARNT. The AHR/ARNT heterodimers act as transcriptional regulators, binding to AHR response elements. 6 It is believed that TCDD activation of AHR/ARNT causes most if not all of the teratogenic effects of TCDD. 6
Retinoids are essential for embryonic pattern formation and organogenesis, and either RA excess or deficiency can disrupt heart development. 7 All-trans RA is an active metabolite of vitamin A that activates the RAR nuclear receptor. 7 RA affects many aspects of heart development, including precardiac field formation, 8 myocyte differentiation,9,10 left-right asymmetry acquisition,11,12 and chamber morphogenesis.9,13 While much work has focused on the effects of RA exposure during the period of early heart development, RA exposure after this period is less studied and produces a phenotype that is very similar to that produced by TCDD.5,13
The receptors for both TCDD and RA are transcriptional regulators. Recently it has become possible to isolate hearts from relatively large numbers from zebrafish embryos. 14 This has made it possible to identify genes that are altered in the heart as TCDD activates AHR. 3 This identified the previously characterized AHR battery of transcripts and numerous other genes altered over time as cardiotoxicity progresses. We also used this technique to determine whether TCDD and RA might alter some gene in common to cause the observed cardiotoxic responses. Time course experiments showed that while the two agents changed few if any transcripts in common at early times, when it might be presumed that the receptors were directly affecting transcription. However, at later time points as signs of heart failure were being manifested, we saw a common cluster of genes down regulated by both agents. This similar transcriptional response/signature was composed of genes involved in cell cycle progression, chromosome assembly, and DNA replication that we called the cell cycle gene cluster (CCGC). 5
Cardiotoxicity as a common response to mutations and chemicals
Anecdotal and published reports suggest that many types of chemical exposure and genetic changes can lead to similar syndromes of altered heart morphology, edema, and circulatory collapse. We hypothesized that if zebrafish embryos are exposed to environmental insults during a critical period in heart development, this can trigger a common response that culminates in heart failure, edema, and circulatory collapse.
To test this hypothesis, we determined whether exposure of zebrafish embryos to a set of unrelated chemicals and morpholino oligonucleotides (MOs) could cause similar patterns of cardiotoxicity. We found that all of the treatments in our test group produced a phenotype characterized by altered heart morphology, massive pericardial edema, and heart failure. In addition, all of these treatments produced the downregulation of a specific set of genes required for cell cycle progression. This downregulation of the CCGC preceded a halt in cardiomyocyte proliferation in all cases. These results indicate the existence of a critical period in fish heart development during which a variety of environmental perturbations can trigger a fatal loss of circulation. Experiments specifically targeting cardiac contractility showed that loss of contraction is sufficient to produce the entire response.
Materials and Methods
Zebrafish embryos
All zebrafish embryos were kept at 27°C in lightly buffered water (60 μg/mL Instant Ocean Salts; Aquarium Systems, Mentor, OH) with a 14-h/10-h light/dark cycle. Cmcl2::GFP embryos were used for heart extraction for microarray analysis. 3 Albino embryos were used for some imaging experiments to avoid situations in which the heart is obscured by pigmentation.
Chemical treatment
TCDD, (>99% purity; Chemsyn, Lenexa, KS) was kept at −20°C as a 1 μg/mL stock solution in DMSO, and embryos were exposed at 1 ng/mL. All-trans RA (≥98% purity; Sigma-Aldrich, St. Louis, MO) was kept as a 10 mM stock solution in DMSO at −80°C, and embryos were exposed at 1 μM. Carbaryl (Sigma-Aldrich) was kept as a 10 mg/mL stock solution in DMSO at room temperature and embryos were exposed at 10 μg/mL. The vehicle control for RA, TCDD, and carbaryl was 0.01% DMSO, the concentration of DMSO in the diluted stocks. Valproic acid (VPA; Sigma-Aldrich) was kept at room temperature at 0.2 M in ethanol as a stock solution and embryos were exposed at 0.06 mM.
For TCDD exposure, embryos were treated with 1 ng/mL TCDD for 1 h at 72 hpf. Embryos were continuously exposed to RA under subdued lighting starting at 72 hpf, with daily changes of solution. Embryos were continuously exposed to VPA starting at 24 hpf with daily changes of solution. Embryos were continuously exposed to carbaryl, starting immediately after fertilization, with daily changes of solution.
Morpholino injection
All MOs were obtained from Gene Tools, LLC (Philomath, OR). Prior to embryo injection, morpholinos were diluted in 1× Danieau's solution [58 mM NaCl, 0.7 mM KCl, 0.4 mM MgSO4, 0.6 mM Ca(NO3)2, and 5 mM HEPES, pH 7.6] as described by Nasevicius and Ekker. 15 MOs were fluorescein tagged at the 3′ ends to monitor injection success.
The TBX5 MO (5′-GAA AGG TGT CTT CAC TGT CCG CCA T-3′) was designed to block the TBX5 translational start site, and was used as a 0.6 mM injection stock. The troponin T2 (TNNT2) MO (5′-CAT GTT TGC TCT GAT CTG ACA CGC A-3′) was designed to block the translational start site of zebrafish cardiac troponin 2 mRNA and used as a 0.16 mM injection stock. The standard Gene Tools Control MO (5′-CCTCTTACCTCAGTTACAATTTATA-3′) was used to control nonspecific responses. Cmlc2::GFP embryos were injected at the 1-cell stage with 3 nL of MO solution per egg.
Zebrafish heart extraction
Zebrafish cmlc2::GFP embryos were anesthetized with Tricaine-S (Aquatic EcoSystems, Apopka, FL), collected in a 1.5 mL microcentrifuge tube, and immersed in 1 mL Lebovitz's L-15 media/10% FBS. Embryonic hearts were separated from the body by sheer force generated by repeatedly drawing and repelling the embryos through a 19 gauge syringe needle connected to a 10 mL syringe, followed by size-fraction of the disrupted embryos.3,14 The hearts expressing GFP were manually retrieved under epifluorescence using a Leica MZ16 stereomicroscope with a pipette, snap frozen in liquid nitrogen and stored at −80°C for further analysis.
Microarray analysis and data processing
For each treatment, three replicate sets of treated and control heart samples were collected for microarray analysis. Samples were collected at 12 h prior to the appearance of outright heart failure and the appearance of severe pericardial edema. This time varied with treatment: 84 hpf for TCDD; 84 hpf for RA; 72 hpf for VPA; 54 hpf for carbaryl, and 72 hpf for the TBX5 MO. Total RNA (around 100 ng) was extracted with Qiagen RNeasy Micro kit from 30–60 hearts (Qiagen, Valencia, CA) for each sample. Biotinylated cRNA was generated from total RNA by Affymetrix Two-cycle Target Labeling and Control Reagents according to the manufacturer's protocol. Fragmented cRNA was then hybridized to Affymetrix GeneChip Zebrafish Arrays, and the arrays were stained and then scanned in an Agilent Gene array Scanner. Scanned .cel images were preprocessed by the GC-RMA function in ArrayAssist Express Version 4.0 software (Stratagene, La Jolla, CA). The output signals were then log2 transformed, and transcripts for which average values were changed by at least twofold in the treated samples compared to controls were selected. For these transcripts the fold change in transcript level produced by the treatment (signaltreatment/signalcontrol) was calculated and the fold change was log2 transformed.
To identify patterns of similarity and difference between the responses to the different treatments we used several clustering algorithms. These included Eigenvalue clustering, Hierarchical Clustering, and Self-organizing Maps. Each method identified a distinct cluster of downregulated transcripts that was largely invariant in composition between the different methods. A set of downregulated transcripts that was clustered together using all three methods was selected, and further filtered to select only transcripts that were downregulated by at least 1.5-fold with all five treatments.
Whole-mount immunohistochemistry and confocal microscopy
Zebrafish cmlc2::GFP embryos were fixed in 4% paraformaldehyde at room temperature for 2 h, followed by bleaching in 6% H2O2 for 2–4 h. Fixed embryos were then stained overnight at 4°C by 1:20 dilution of rabbit anti-phosphohistone H3 (Ser 10) (sc-8656-R) and Alexa Fluor 488 conjugated anti-GFP antibody (sc-9996) (Santa Cruz Biotechnology, Santa Cruz, CA), followed by washing and secondary antibody probing with Alexa Fluor 546-conjugated secondary anti-rabbit antibody (A11071; Invitrogen, Eugene, OR) for 2 h at room temperature. Embryonic hearts were then imaged under a Nikon C1 laser scanning confocal microscope (Nikon, Inc., Melville, NY) by Z-disk imaging with the section thickness at 1.4 μm. Total number of proliferating cardiomyocytes was counted and summed from each image of the 61 Z-disk sections for each respective experiment.
Results
Different stressors produce a similar pattern of heart failure in developing zebrafish embryos
Previous work in our laboratory has shown that TCDD and RA, two cardiac teratogens with unrelated mechanisms of action, elicit a similar pattern of heart failure in zebrafish embryos.3,5 This syndrome is characterized by an elongated heart of reduced size, circulation failure, and severe pericardial edema. 4
Anecdotal evidence has suggested that a variety of insults, including environmental stresses and many different mutations often produce a type of heart failure, which is similar to that produced by TCDD. We postulated that these defects are in fact more than superficially similar, involving an as yet unrecognized common process.
To test this hypothesis we compared the effects of a set of stressors on embryonic heart development. These agents matched two criteria: they are known to affect zebrafish heart development, and they are thought to act through different mechanisms, affecting distinct cellular pathways. This group included TCDD, RA, VPA, carbaryl, and MO knock down of TBX5 expression.
TCDD acts through the AHR/ARNT transcription factor. 4 RA is an agonist for the RAR/RXR nuclear receptor. 7 VPA is an anticonvulsant used to treat epilepsy and is also known to affect cardiac development in zebrafish embryos. 16 Carbaryl is an acetylcholinesterase inhibitor pesticide that causes defective cardiac morphology in medaka and zebrafish embryos.17,18 The TBX5 gene plays an important role in cell-type specification and morphogenesis, and is crucial for heart development. 12
Despite these differences, all of these agents produced a similar response: an elongated heart with a thin atrium, a compacted ventricle, and massive pericardial edema (Fig. 1). These morphological defects were associated with the development of circulatory collapse. In contrast, the hearts in control animals exposed to vehicle maintained compact chambers and showed no signs of heart failure or edema.
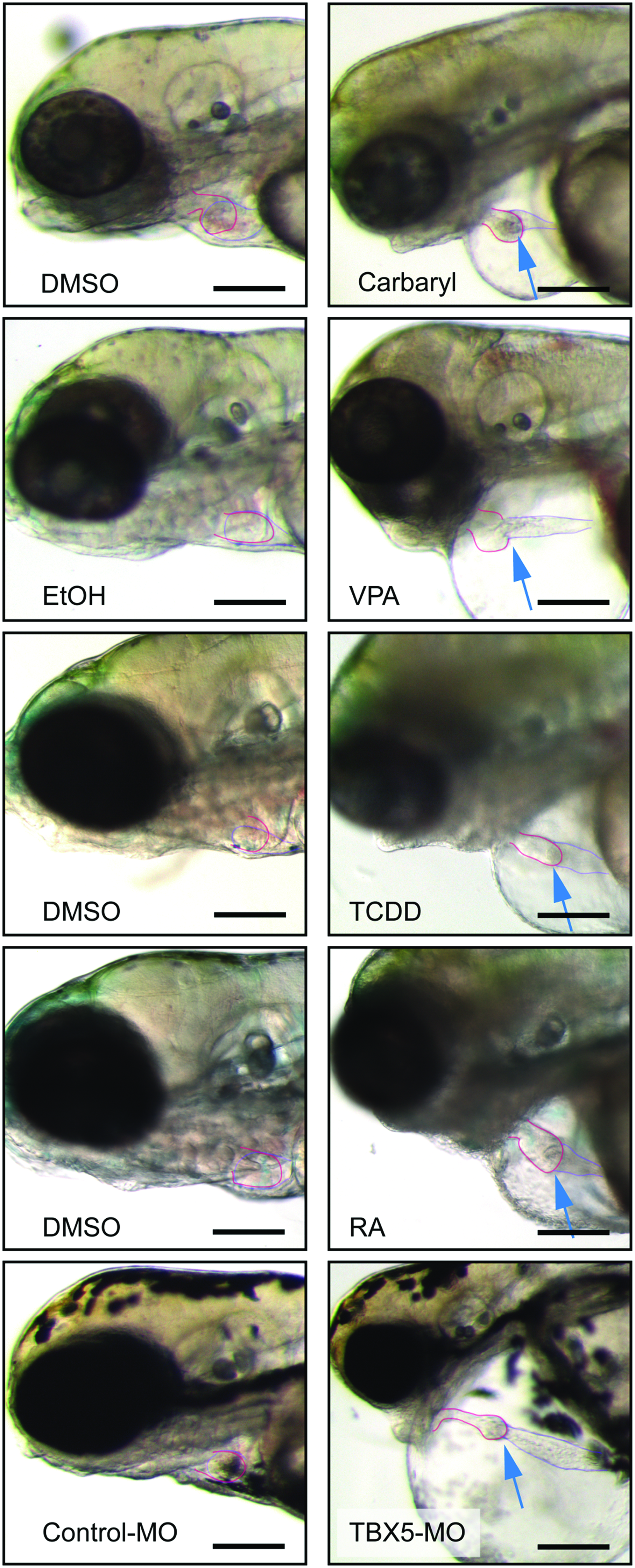
Multiple different treatments produce similar cardiotoxicity in zebrafish embryos. Zebrafish embryos were exposed to carbaryl, VPA, TCDD, RA, and TBX5-MO as described in the Materials and Methods section. Fish used as controls are shown to the left for each treatment. Carbaryl-treated fish were photographed at 72 hpf, VPA, and TBX5-Mo-treated fish were photographed at 96 hpf, and the TCDD and RA fish are shown at 120 hpf. Arrows point to the heart. The atria and ventricles are outlined. Dark scale bar illustrates a 100 μm length. TCDD, tetra-chlorodibenzo-p-dioxin; RA, retinoic acid; VPA, valproic acid; MO, morpholino oligonucleotides. Color images available online at www.liebertpub.com/zeb
It should be noted that it was necessary to make variations in the dosing time course to adjust for the properties of each compound. For example, TCDD is lipophilic and stable to degradation. Therefore, a single 1 h exposure to TCDD at 72 hpf leaves behind a continuous body burden of TCDD, and produces the same cardiovascular defects as would be seen with continuous exposure immediately following fertilization. 3 In contrast, RA is unstable in bright light and is metabolized in the body, so embryos were continuously exposed to RA with daily renewal of solution beginning at 72 hpf. MOs can only be injected at the one cell stage if they are to be evenly distributed across the dividing cells, so the TBX5 MO was injected immediately after fertilization. Carbaryl was most effective in causing cardiotoxicity when exposure began immediately after fertilization, and VPA was most effective when exposure began at 24 hpf. Nonetheless, all of these treatments produced similar cardiovascular collapse.
A common transcriptional response to a group of unrelated stressors
Microarray experiments shows that five treatments of TCDD, RA, VPA, carbryl, and TBX5-MO knockdown cause the downregulation of a very specific set of genes as heart failure progresses. This set of transcripts is associated with processes needed for growth: 76.3% of the mRNAs comprising this set encode proteins needed for cell cycle progression. We have termed this group of genes the CCGC. CCGC was downregulated by each treatment, despite the fact that these treatments produced otherwise quite distinct patterns of transcript changes (Fig. 2).
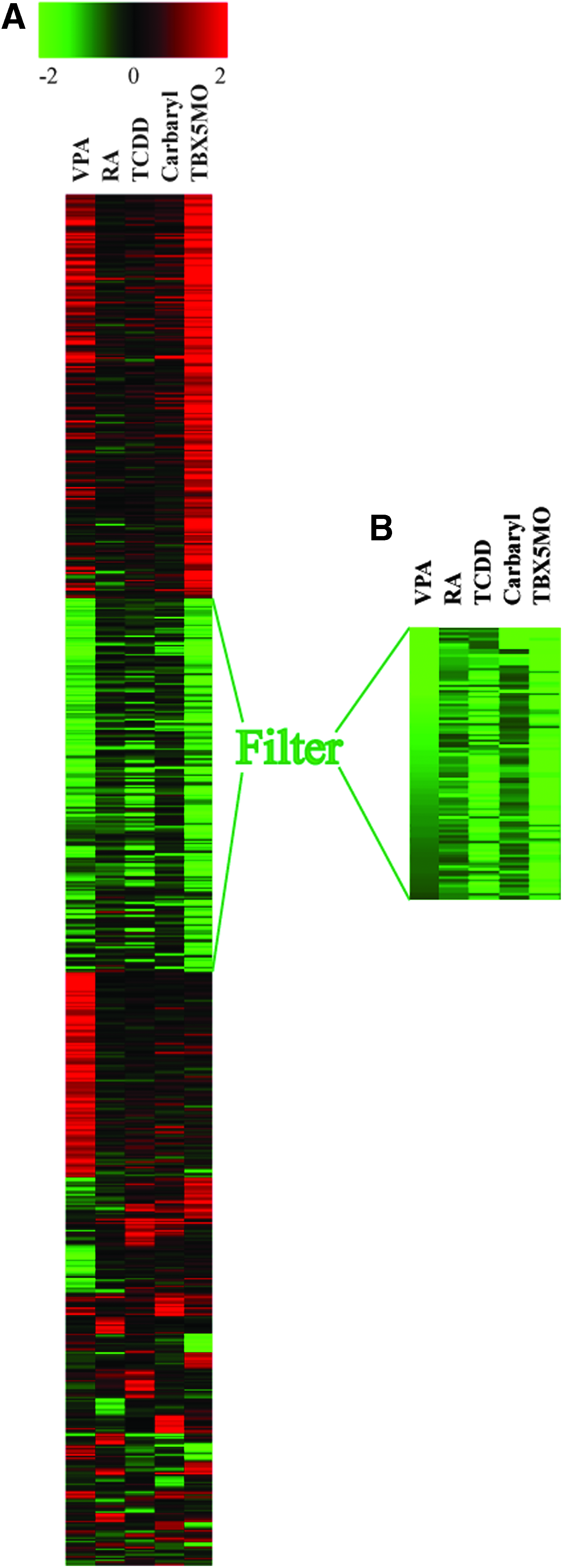
Multiple different treatments produce a common signature of downregulated cell cycle genes in the developing embryonic heart. Embryonic zebrafish were treated with carbaryl, VPA, TCDD, RA, and TBX5-MO and heart samples were collected for microarray hybridization as described in the Materials and Methods section.
For each treatment group, we collected samples at a point ∼12 h prior to circulatory collapse and heart failure for array hybridization. For each treatment, we selected those transcripts that were altered by at least twofold compared to its control. We then combined the data sets to include all transcript affected by twofold or more in at least one of the treatments. The expression data for each of these transcripts were compared across all experimental treatments by K-means clustering algorithm. A set of downregulated transcripts is identified in the heat map (Fig. 2A). This clearly shows a set of downregulated genes common to all of the treatments, while the remaining transcript changes appear to be specific to a given individual treatment.
To obtain a set of genes representing a core cluster of genes downregulated by the different treatments, we selected transcripts that clustered together in five different clustering runs using self-organizing Map, K-means, and Eigenvalue clustering (see experimental procedures) to generate a gene cluster composed of 92 transcripts that were consistently grouped together, downregulated by at least twofold in one or more of the treatments, and by at least 1.5-fold by all treatments (Table 1). The functions of the genes within this cluster were queried using Affymetrix NetAffy and Ensemble annotation. Of this set, 76.3% of the transcripts function in chromosome assembly/cell cycle progression/DNA replication (Fig. 2B). We consider this list (Table 1) to be a refinement of the CCGC previously identified by RA and TCDD as sole stressors. 5
Notably, we collected samples for microarray analyses at time points prior to the appearance of obvious heart failure. Thus, the downregulation of CCGC is associated with and precedes the occurrence of heart failure and circulatory collapse. We postulate that the CCGC might be useful biomarkers associated with heart failure, and that downregulation of transcripts within the CCGC might directly contribute to heat failure.
Different agents halt cell proliferation in the heart
We have previously shown that TCDD halts heart cell proliferation prior to outright signs of heart failure. This is consistent with the downregulation of CCGC transcripts. To determine whether this is common to all of the agents tested in this report, we used immunohistochemistry with anti-phosphohistone H3 antibodies to identify proliferating cells in the hearts of developing zebrafish treated with TCDD, RA, VPA, carbaryl, and the TBX5-MO. To be certain that we properly identified proliferating heart cells, we used a strain of zebrafish expressing GFP from the cmlc2 promoter, and antibodies recognizing GFP. With this method, anti-phosphohistone H3 produces a red nuclear signal, while the antibody recognizing GFP stains the cardiomyocytes green. Confocal merging producing a yellow signal indicates a proliferating cardiomyocyte (Fig. 3).
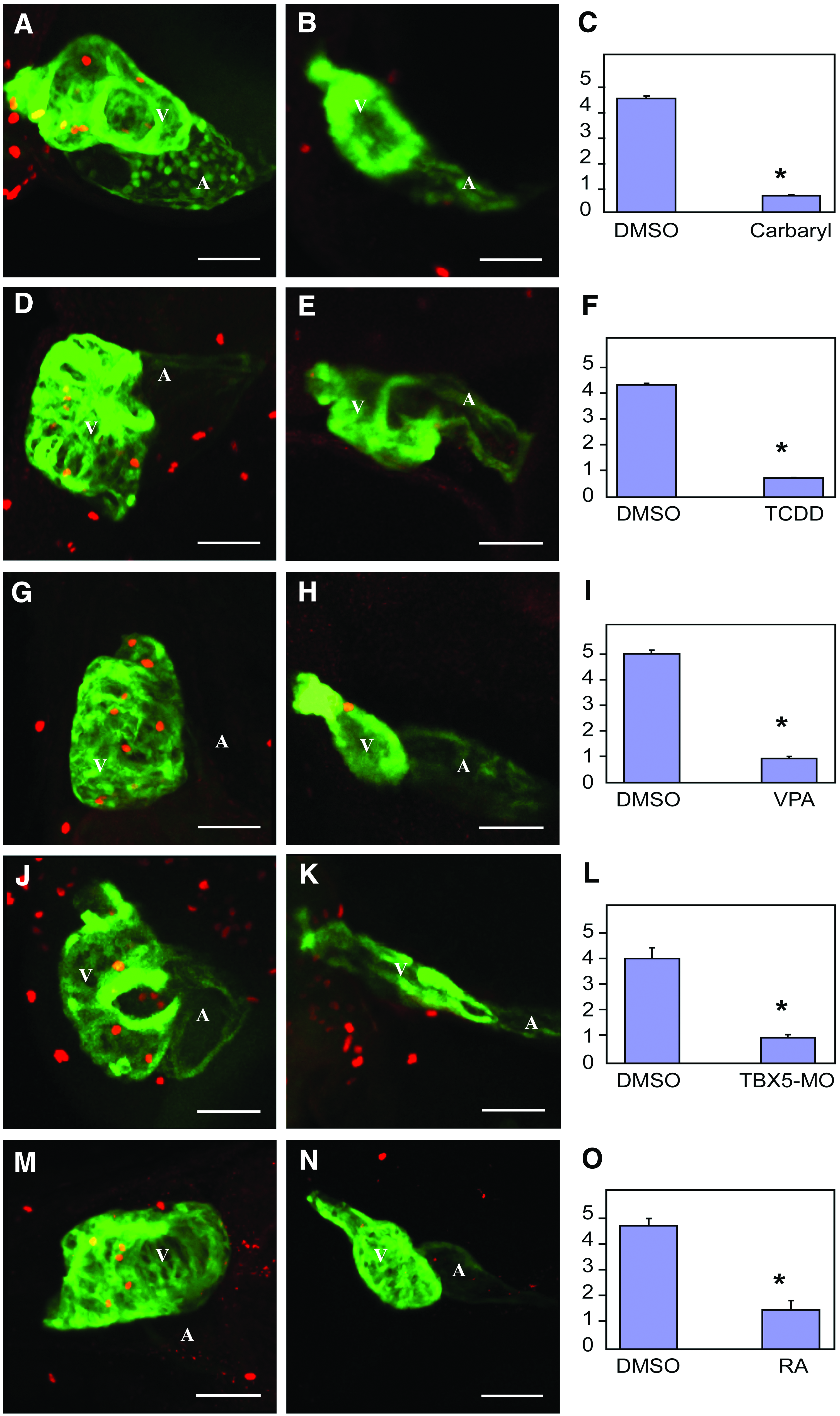
Multiple different treatments produce a halt in cardiomyocyte cell division. Zebrafish embryos were exposed to carbaryl, VPA, TCDD, RA, and TBX5-MO as described in the Materials and Methods section. Embryos expressing GFP in cardiomyocytes were fixed for immunohistochemistry with anti-phosphohistone H3 and anti-GFP and imaged by Z-disk confocal microscopy as described in the Materials and Methods section. Cells with both anti-PH3 and anti-GFP staining were revealed as cardiomyocytes. Three replicate experiments were conducted for each treatment. For each replicate, 4–10 embryonic hearts were examined. The average number of proliferating cardiomyocytes for each treatment is indicated by a bar graph.
Confocal microscopy revealed that there were consistently fewer proliferating cardiomyocytes within the hearts treated with TCDD, RA, VPA, carbaryl, or the TBX5-MO (Fig. 3B, E, H, K, N). We postulate that the downregulation of the CCGC transcripts plays a role in halting cardiomyocyte proliferation in these hearts.
Loss of contractility as a cause of heart deformation and failure
Carbaryl, VPA, TCDD, RA, and TBX5-MO knockdown act through different routes, yet produce surprisingly similar effects on the developing heart. Anecdotal reports indicate that many different zebrafish mutations produce similar effects associated with heart malformation and pericardial edema. This suggests that some sort of physiological cycle is triggered that leads to heart failure.
In studying TCDD toxicity, we noted that TCDD produces a decrease in stroke volume several hours prior to the decrease in CCGC mRNAs.3,4 This suggests the possibility that decreased cardiac output triggers CCGC repression and the cell cycle halt.
To test this hypothesis, we chose a very specific method of reducing cardiac contractility. We injected eggs with MOs directed against the product of the silent heart gene, cardiac TNNT2. Cardiac TNNT2 is a component of the muscle fibers, regulating tropomyosin activation in response to calcium signals. 17 Therefore, hearts in the TNNT2 morphants cannot beat in response to the action potential. 18 TNNT2 is not thought to be a developmental regulator; instead TNNT2 is a part of the structural apparatus that produces muscle contraction. Therefore, one might expect the TNNT2-MO to produce zebrafish carrying a heart that is normal in appearance with no contractions. On the other hand, if as we proposed above, a loss of contractility produces heart elongation and pericardial edema, then the TNNT2-MO should produce very similar effects on heart morphology. Indeed, the TNNT2-MO-injected zebrafish embryos phenocopied the heart failure produced by the other treatments completely (Fig. 4).
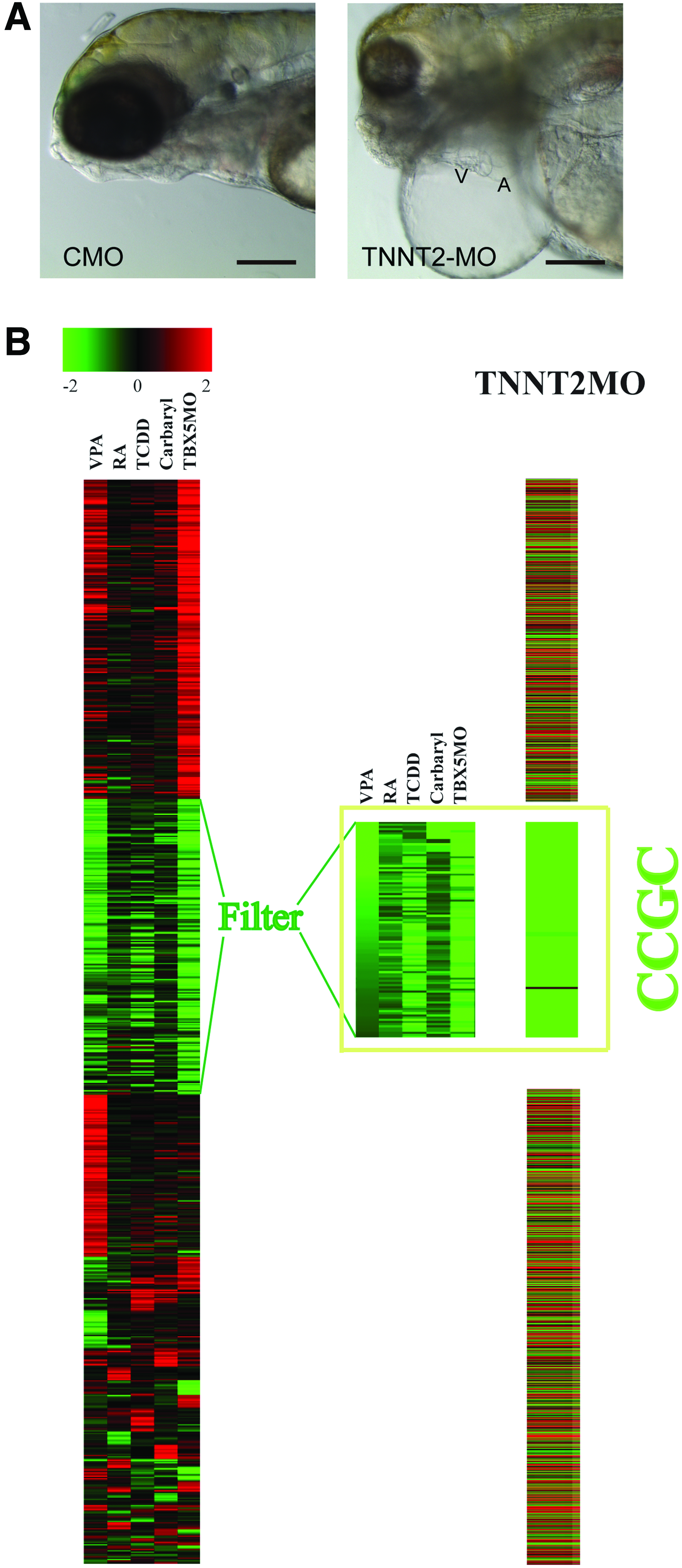
MO knockdown of zebrafish TNNT2 causes downregulation of the CCGC.
By 120 hpf, the TNNT2 morphants had developed elongated hearts along with severe pericardial edema (Fig. 4A), in contrast to the compact hearts observed in zebrafish receiving the control-MO. Thus, proper cardiac contractility and the accompanied correct blood flow are crucial for maintenance of normal embryonic heart shape.
Loss of contractility triggers downregulation of CCGC. The morphological effects of the TNNT2-MO are somewhat general and non-distinct; it is difficult to be certain that the edema and elongated heart produced by one treatment exactly matches the edema and elongated heart produced by another. It is therefore difficult to determine whether the different treatments are indeed triggering the same response in the developing heart based on the morphological data alone. However, the downregulation of the CCGC is a very specific and distinct response shared by TCDD, RA, TBX5-MO, carbaryl, and VPA treatment. If blocking cardiac contractility is truly causing the same effect as these agents, then the TNNT2-MO should also produce a downregulation of this set of genes. The chance of this occurring by random chance is exceedingly remote.
We used microarray experiments to test this. Measurement of transcript levels in the hearts of TNNT2 morphants showed that while many of the transcript changes produced by loss of TNNT2 were unique to the treatment, practically every member of the CCGC was reduced in comparison to those in control hearts (Fig. 4B).
As with the other treatments, the TNNT2-MO also caused a halt in cardiomyocyte proliferation, bringing heart muscle cell division to a halt (Fig. 5). Immunohistochemistry using anti-phosphohistone H3 antibody staining along with staining specific for cardiomyocytes showed that while control heart cells continued to divide the dividing cardiomyocytes in TNNT2 morphants were almost entirely absent. Therefore, a very specific mutation that blocks the contraction of heart muscle fibers triggers a process that leads to changes in heart morphology, edema, and a very specific change in gene expression associated with cell cycle arrest. This exact suite of responses is also produced by a variety of different stressors acting through a wide range of mechanisms.
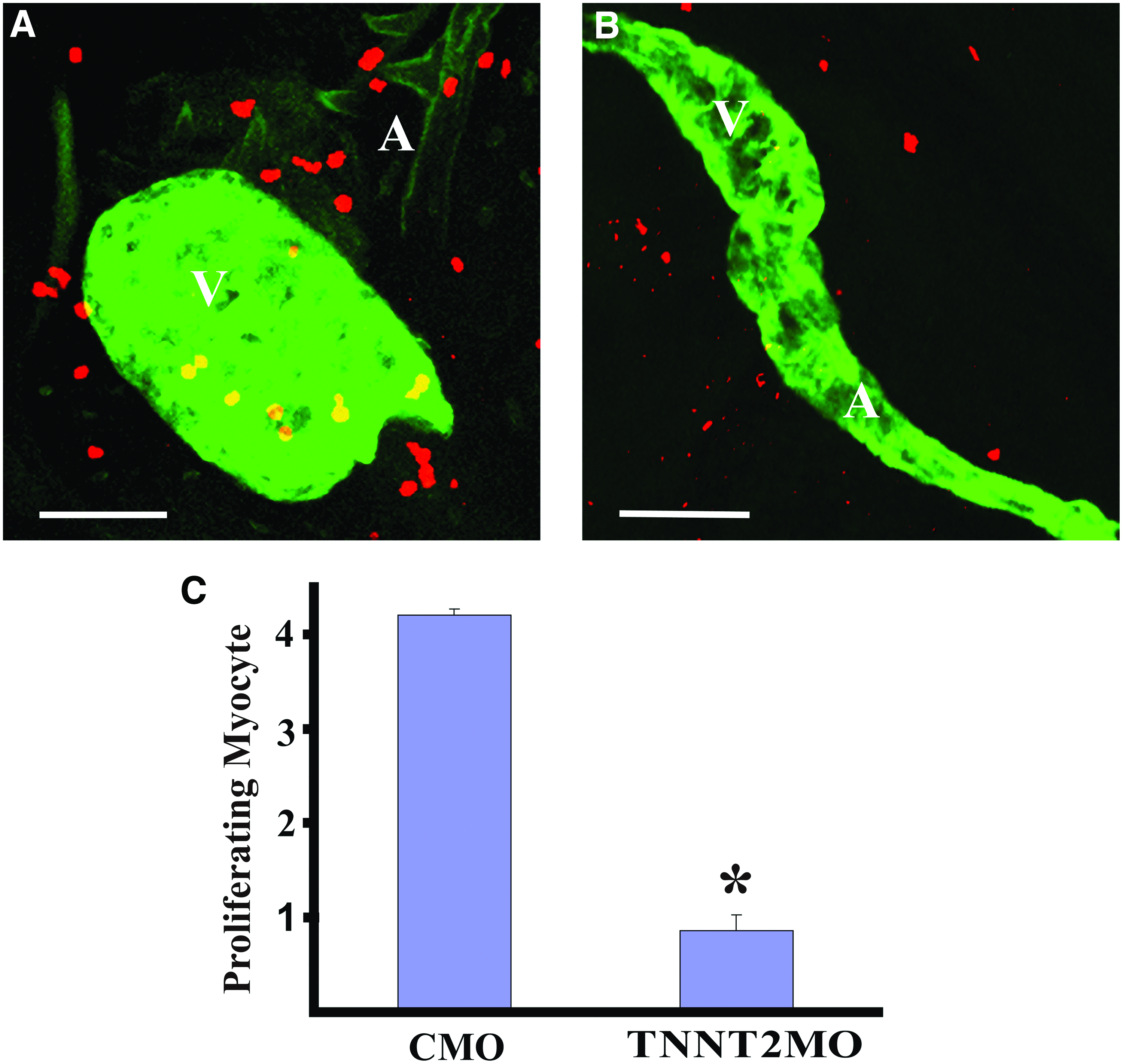
MO knockdown of zebrafish TNNT2 halts cell division in cardiomyocytes. Zebrafish were injected with either the control-MO (CMO) or the TNNT2-MO, and treated for immunostaining and confocal microscopy as described for Figure 3. The proliferation of cardiomyocytes was also reduced in the hearts of TNNT2 morphants.
Discussion
A common pathological response produces zebrafish end point heart failure
We repeatedly observed that developing zebrafish embryos exposed to a variety of different chemical and genetic insults died with end-stage heart failure. These agents ranged from the prototypical AHR agonist, TCDD, to an inhibitor of acetylcholinesterase, carbaryl, to a morpholino against TBX5. While there is no reason to suppose that these agents should not affect the heart, there is no a priori reason to think that these should produce such a similar set of responses at the heart. Each of these treatments produced a feeble, elongated heart surrounded by a pericardium distended by edema. Further, as the heart failed, a common set of genes promoting cell proliferation was repressed. While cells in surrounding tissues continued to divide, the myocardial cells ceased division. Our results are consistent with the anecdotal observations that many different mutations affecting one or another property of the heart tend to produce exactly this phenotype of pericardial edema and elongated heart. This suggests that during a certain period of development the zebrafish is especially vulnerable to agents that trigger this series of events.
A molecular signature associated with the heart failure response
We adopted a microarray strategy to analyze the transcriptional profile of these hearts at timepoints prior to overt failure. Pattern recognition software identified a common cluster of transcripts known to be involved in chromosome assembly, DNA replication, and cell cycle progression that were downregulated in these failing hearts. This molecular signature is consistently associated with and precedes failure.
It is unclear what triggers downregulation of the CCGC. Coregulated genes sometimes share common regulatory modules.21,22 We postulate that there might be a potential common set of transcription factors regulating the downregulation of CCGC. 23 Various computer algorithms can be used to search for such transcription factors. CLOVER (Cis-element OVERrepresentation) (http://zlab.bu.edu/clover/) 24 is an algorithm that can be used to search for transcriptional factor binding motifs enriched in a set of coregulated DNA sequences. CLOVER uses the JASPAR CORE library composed of known transcription factor motifs. 25 In our study, 14 enriched motifs in the 5kb regions upstream of the transcription start sites of these CCGC genes were identified. These motifs include NF-Y CAAT-BOX, Foxd1 FORKHEAD, C2H2 ZN-FINGER, E2F1 and others (Table 2). Among these motifs, C2H2 ZN-FINGER has the highest hits with a raw score of 101. However, this group of transcription factors is extremely common in vertebrates, 26 which undermines its importance possibly specified as a master regulator for CCGC downregulation. The second highest hit, the NF-Y complex, made of three subunits, A, B, and C, is a CCAAT box binding factor. 27 NF-Y is found in the promoter regions of cell cycle related genes, 28 and it is reported to maintain the promoter activity of cyclin B1, cyclin B2, and cdc2,28,29 which are among the list of CCGC members. Identification of transcriptional regulators may be one method for understanding the mechanism that causes this heart failure response, linking mechanical failure as a cause in perturbed local gene expression to pathological structure formation.
Clover program identified the enriched motifs within the 5 kb 5′ regions upstream of CCGC gene transcription start sites, using all 5 kb 5′ regions of the transcription start sites of all known zebrafish genes as a background control. Statistical significance of a given motif in a set of regulatory sequences is described as p, with a value<0.01 indicating the significance of enrichment of this motif in these target sequences.
CCGC, cell cycle gene cluster.
Multiple stressors converge to halt cardiomyocyte proliferation
Under normal conditions, cardiomyocytes are proliferating in the zebrafish heart at the stages investigated until the juvenile stage. The total number of cardiomyocytes increases during this period, and immunostaining shows both BrdU incorporation and histone H3 phosphorylation in cardiomyocytes at 72 and 120 hpf.3,5 On the other hand, all of the treatments examined repressed CCGC transcripts and halted cardiomyocyte division. Thus, we postulate that the decrease in heart size may be partly due to the decreased cardiomyocyte proliferation in the stressor-treated hearts. Cardiomyocyte proliferation significantly contributes to the increasing number of myocytes in the embryonic heart leading to its growth. In juvenile zebrafish, it is clear that the proliferation of a small subset of clonally dominant cardiomyocytes contributes to the increase of size of the heart during its growth. 30 In embryos, the hypothesis of an alternative source for supplying cardiac progenitor cells from the epicardium was denied by a lineage-tracing study, confirming the possibility that the mechanism for embryonic heart growth is similar to that of juveniles. 31
What causes the heart failure response?
Various stressors, with divergent mechanisms, all cause a similar phenotype. The agents were deliberately chosen to cover a wide range of target pathways, yet they produced a common pathological signature of impaired cardiac function and blood flow. Anecdotal evidence suggests that many other stressors and mutations also initiate a process that ends in heart failure. How do these agents converge to produce this response?
One clue comes from our earlier experiments with TCDD. The earliest alteration in heart function that we observed was decreased ventricular stroke volume, occurring several hours before either edema or the decrease in CCGC levels. It is thought that changes in hemodynamics affect embryonic heart development. 32 This led us to the idea that TCDD, and perhaps the other agents, causes an initial effect on heart function that then triggers some previously unrecognized response in tissue gene expression that leads to the alterations in heart morphology, edema, and cell cycle arrest that we observed.
An obvious test of this hypothesis is to specifically block myocardial contractility. If the hypothesis is true, then this should produce all of the responses described above. If the hypothesis is not true, then we should have seen an otherwise normal looking heart with no contractions.
We used MOs to knock down cardiac troponin T, a crucial component of the contraction apparatus that functions as a calcium sensor, bridging the action potential with cardiomyocyte contraction. In zebrafish embryos, loss of function mutations in cardiac TNNT2 led to a silent heart without rhythmic contraction. In our experiments loss of TNNT2 also produced a gross alteration in heart shape, pericardial edema, and a downregulation of a specific set of cell cycle genes. All of these responses were shared with the other agents.
Clearly, developing zebrafish embryos as a heart failure model for probing the relationships between pathological hemodynamics, transcriptome, and structure malformation does not need a self-destruct mechanism built into the developing heart. Yet, our results suggest that such a response exists. It is noteworthy that in human heart failure the inappropriate triggering of normal homeostatic responses often plays a part in a downward spiral that worsens the disease. The heart failure that we see in the developing zebrafish may also involve normal responses that in some circumstances contribute to a downward spiral in cardiac function.
We propose that these chemicals reveal a period in which the developing heart is extremely sensitive to perturbations; especially perturbations in blood flow and pressure. It is thought that cardiac function and the resulting flow of blood through the heart contributes to the formation of cardiac shape. Regionally confined cell shape changes are influenced by cardiac function and blood flow, producing a major contribution to cardiac shape. 32 Changes in gene expression or signaling pathways induced by endothelial sheer forces may also be factors contributing to the changes in heart shape.33,34 This means that any agent that alters cardiac output during this time can trigger changes in heart morphology. If these changes further reduce cardiac output, a positive feedback loop, or viscous cycle, is produced. This would explain how different agents could produce a response that looks the same at the conclusion.
Significance
Does this mean that each compound produces a set of nonspecific responses? The answer is no, for two reasons. First, while the end-stage heart failure with full-blown edema and elongated heart may be triggered by a variety of agents, each agent did trigger the response in different ways. This means that the agents must in some way alter heart function at the beginning. The resulting catastrophic heart failure is an amplified response to that initial insult. While the response at the very end is very similar, the initiation of the response is likely to involve a specific process for each compound.
Second, regardless of the specificity of the mechanism, this response is lethal in early life stage fish. Thus, from a practical standpoint, compounds that initiate this response are a danger to fish populations if exposure occurs during development. Further, we cannot say that the mechanisms acting in zebrafish do not also occur in vertebrates such as humans. Thus, our results may provide insight into mechanisms underlying human congenital heart disease. Congenital heart disease in some case is associated with decreased ventricular mass termed as ventricular hypoplasia. For instances, in pulmonary atresia or tricuspid atresia, the lack of blood flow in the right side of the heart leads to a small right ventricle, 35 where the mechanism we proposed might play an important role. Thus it is possible that the effects we see in zebrafish have parallels in human disease for understanding the interplay between organ function, gene expression and structure.
Footnotes
Acknowledgments
We thank Craig A. Struble from Marquette University, Milwaukee, Wisconsin, for his assistance in genome-wide computational work to identify the enriched transcriptional factor recognition motifs by CLOVER program. This work was supported by the National Sciences Foundation of China grant (30971106, to J.C.), and the grant from the Scientific Research Foundation for Returned Overseas Chinese Scholars of Education Ministry of China ([2010] 1561, to J.C.).
Disclosure Statement
The author declares no conflict of interests.