
Abstract
Abstract
Research on common carp, Cyprinus carpio, is beneficial for zebrafish research because of resources available owing to its large body size, such as the availability of sufficient organ material for transcriptomics, proteomics, and metabolomics. Here we describe the shot gun sequencing of a clonal double-haploid common carp line. The assembly consists of 511891 scaffolds with an N50 of 17 kb, predicting a total genome size of 1.4–1.5 Gb. A detailed analysis of the ten largest scaffolds indicates that the carp genome has a considerably lower repeat coverage than zebrafish, whilst the average intron size is significantly smaller, making it comparable to the fugu genome. The quality of the scaffolding was confirmed by comparisons with RNA deep sequencing data sets and a manual analysis for synteny with the zebrafish, especially the Hox gene clusters. In the ten largest scaffolds analyzed, the synteny of genes is almost complete. Comparisons of predicted exons of common carp with those of the zebrafish revealed only few genes specific for either zebrafish or carp, most of these being of unknown function. This supports the hypothesis of an additional genome duplication event in the carp evolutionary history, which—due to a higher degree of compactness—did not result in a genome larger than that of zebrafish.
Introduction
In addition to physiological comparisons between zebrafish and carp, a comparison of their genomes can give interesting information on vertebrate evolution. Two rounds of whole genome duplications occurred at about the time of the radiation of the early vertebrates, whereas another genome duplication occurred at the base of teleost radiation. The study of genomes of several teleost species already has given new insights into the amount of genetic data that has remained after this duplication. For example, our recent publication on the European eel genome shows that this fish species contains a surprisingly complete set of duplicated Hox clusters, showing that some fish species have been very stagnant in preserving duplicated gene sets. 20 In several groups of fish, an additional tetraploidization took place, an event postulated to be more recent in cyprinids (∼11–21 MYA)21,22 than in salmonids (∼25–100 MYA). 23 In contrast with salmonids, polyploid cyprinids generally do not show chromosome quadrivalents, but have their chromosomes consistently arranged in bivalent pairs during meiosis, supporting an allotetraploidization origin. In the cyprinid lineage, zebrafish (2n=50) and tetraploid common carp (2n=100) diverged some 30 million years ago. 24 The polyploidy status of common carp, but also goldfish (Carassius carassius), is supported by a karyotype consisting of approximately 100–104 chromosomes, as well as more and larger erythrocytes than their diploid relatives.25,26 However, the DNA content per haploid genome is similar for carp and zebrafish, with estimates ranging from 1.61 to 2.03 picogram for common carp, and from 1.68 to 2.28 picogram for zebrafish (www.genomesize.com).
A major bottleneck for making optimal use of this combination of model organisms is the absence of genomic sequence data for the common carp. Despite a long history of domestication and transfers, two existing subspecies can be defined as C. carpio carpio (Europe) and C. carpio haematopterus (East Asia). 27 Although recent progress has been made in obtaining sequence data from BAC-end libraries of C. carpio28,29 and preliminary paired-end random sequence data has been described, 30 there are no high coverage genomic sequence data sets present in the data base for any carp species. Since the carp is proposed to be a tetraploid with an estimated number of 100 chromosomes, nucleotide sequencing of non-inbred strains most likely make genome assembly very difficult. We therefore have used double haploid derivatives from a clonal carp line in order to reduce the number of polymorphisms. Using this approach, we obtained high coverage random sequence data of paired-end and mate pair libraries using Illumina Technology. We report on a resulting genome assembly with 33 times coverage of the entire genome and have compared the predicted exome with that of zebrafish.
Materials and Methods
Common carp fish lines and DNA isolation
Genomic DNA was isolated from red blood cells of a homozygous clonal common carp line shown in Figure 1 using the Qiagen Blood and Tissue DNeasy kit according to the manufacturer's description (Qiagen, Hilden, Germany). European common carp (Cyprinus carpio carpio L.) R3×R8 are the offspring of a cross between fish of Hungarian origin (R8 strain) and of Polish origin (R3 strain).26,31 A single sexually mature female (number 69) was taken from this heterozygous base population (R3xR8) for artificial reproduction by induced gynogenesis based on inactivating the paternal genome with ultraviolet light and restoration of ploidy through suppression of the first mitosis with a temperature shock. 32 Following the first gynogenetic reproduction, again a single sexually mature female (number 45) from the now homozygous offspring was reproduced by induced gynogenesis.33,34 This resulted in a homozygous all-female clonal line maintained homozygous by normal fertilization making use of sex-reversed progeny. 17 Genomic DNA was taken from nucleated red blood cells collected from a single individual from this R3R8 69-45 homozygous clonal carp line.
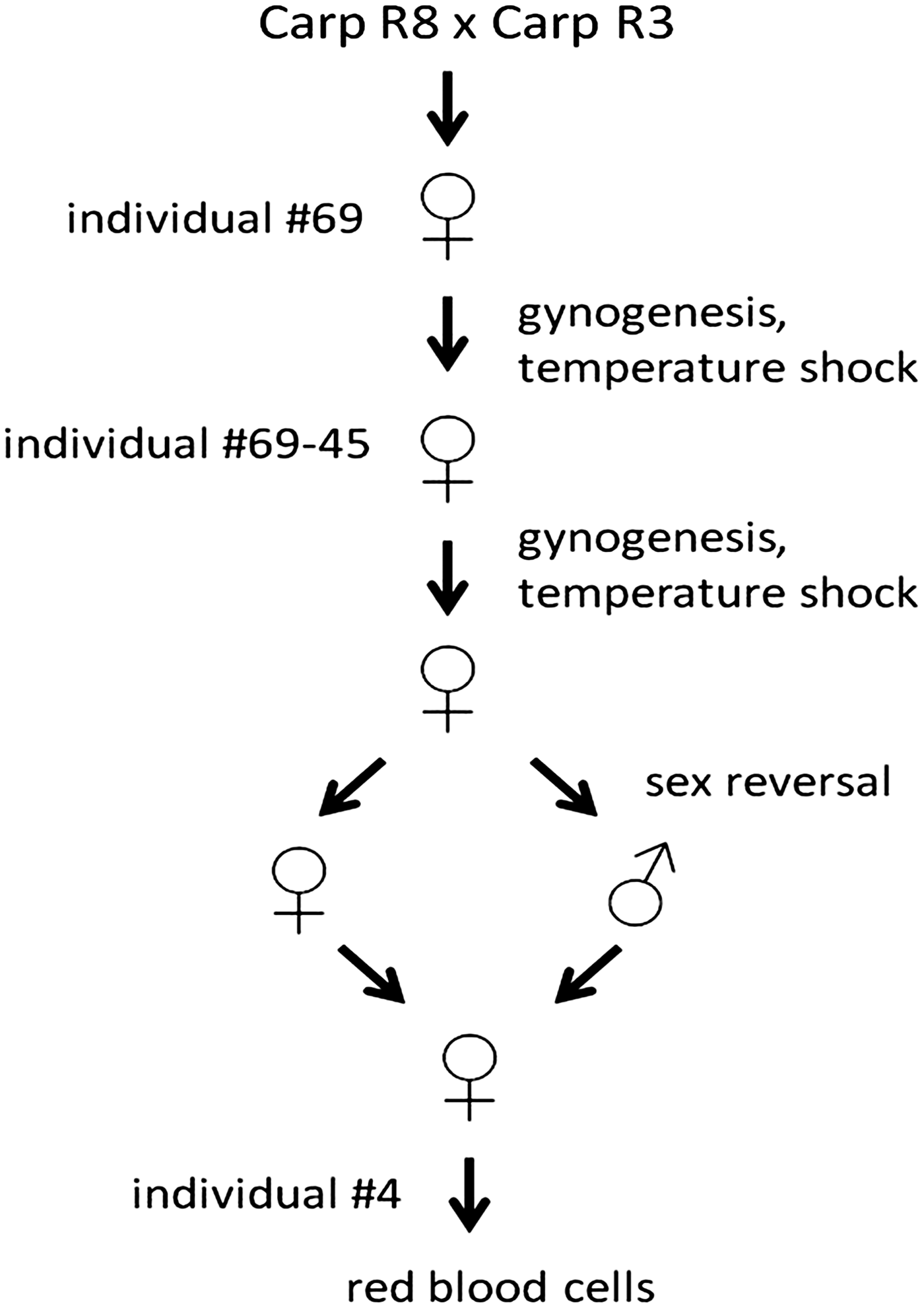
The strategy used to obtain the common carp fish line used for genome sequencing. Details are described in the Methods section. The resulting carp line is available at Wageningen University.
Construction of genomic libraries
Paired-end libraries were prepared from 5 μg of isolated gDNA using the Paired-End Sequencing Sample Prep kit according to the manufacturer's description (Illumina Inc., San Diego CA). Either a 160–200 bp band or a 600–650 bp band was cut from the gel. After amplification for 10 cycles, the resulting libraries were analyzed with a Bioanalyzer 2100 DNA 1000 series II chip.
Mate pair libraries were prepared from 20 μg of isolated gDNA using the Mate Pair Library Prep Kit v2 (Illumina Inc.). Either a 2–3, a 4–5 kb (two individual isolations), a 5–6 kb, or a 6–7 kb band was isolated from gel. After the first gel purification, the fragment length was analyzed using an Agilent Bioanalyzer 2100 DNA 12000 chip. After circularization, shearing, isolation of biotinylated fragments, and amplification, the 400–600 bp fraction of the resulting fragments was isolated from gel. Finally, the libraries were examined with an Agilent Bioanalyzer 2100 DNA 1000 series II chip, and the concentration was determined using qPCR (KAPA Biosciences, Woburn MA).
The following are the insert sizes of the libraries: PE200 library, target insert size=200, average insert size (aligned)=111, 95% of pairs in the 75–152 interval; PE600 library, target insert size=600, average insert size (aligned)=545, 95% of pairs in the 517–587 interval. For the mate pair libraries, the estimates are: 2kb: 1900–2700; 4kb: 3500–6000; 5kb: 4500–7000; 6kb: 5000–9000 (Table 1).
Illumina sequencing
Both genomic paired end libraries were sequenced using an Illumina GAIIx instrument, or a HiSeq 2000 according to the manufacturer's description. Genomic paired-end libraries were sequenced with a read length of 2x76, or 2x151 nucleotides (to ∼33-fold genome coverage).
The genomic mate-pair libraries were sequenced on a HiSeq2000 instrument with a read length of 2x51 nucleotides (to ∼8-fold genome span) according to the manufacturer's description. Image analysis and base calling were done by the Illumina pipeline.
Genome assembly and gene prediction
Sequencing reads from both paired end libraries were used for de novo assembly. Reads potentially contaminated with Illumina adapter sequences were discarded. The frequency of 17–19-mers in the raw data was counted using Jellyfish. 35 Based on these distributions, the haploid genome size of Cyprinus carpio carpio was estimated 36 to be 1.4–1.5 Gbp in size. The distribution of 19-mers was used to identify and correct sequencing errors using Quake. 37
Where possible, read pairs were merged into long single reads. 79% and 1% of pairs could be merged into single fragments with mean lengths of 105 and 211 nt for libraries PE200 and PE600, respectively.
Final preprocessed reads were assembled into contigs using the CLC bio Assembly Cell version 3.2 de novo assembler (CLC bio, Aarhus, Denmark), using a k-mer setting of 31.
Sequencing reads from library PE600 and all mate pair libraries were used to merge the resulting contigs into larger scaffolds. All reads were aligned to the assembly using Bowtie, 38 and potential clonal fragments (the result of low complexity mate pair libraries) were discarded. SSPACE 39 was used to construct scaffolds from contigs linked by at least three non-redundant link pairs.
Over 99% of corrected reads from both paired-end libraries aligned to the assembled contigs (CLC bio reference assembler at default settings), indicating that the assembly is an accurate representation of the genomic sequence sampled by both libraries. In addition, over 99% of corrected PE600 reads aligned to an initial assembly constructed using PE200 reads only (N50=2262 bp), demonstrating that both independent libraries sampled the same random fraction of the genome (and therefore, the entire fraction of the genome suitable for Illumina sequencing).
Augustus 40 was used to predict potential exons and genes on the final scaffolds. RNA deep sequencing datasets were aligned to predicted transcripts, quantified and normalized using the CLC bio Genomics Workbench version 4.9 (CLC bio). The Carp scaffolds were submitted to NCBI as Bioproject Accession: PRJNA73579, currently under review for release by NCBI and is publicly available as a FASTA file at www.carpgenome.com, together with the RNA deep sequencing datasets of carp embryos and adult tissues. For zebrafish and medaka gene predictions, ENSEMBL Zv9 and MEDAKA1 were used, respectively (www.ensembl.org).
Results
Assembly of the common carp genome
Based on blood material from the carp line that were generated following the scheme shown in Figure 1, we have prepared genomic libraries as described in the Materials and Methods section. Illumina sequencing of these libraries resulted in a genome assembly as described in the Materials and Methods section and summarized in Table 2. The approach used is similar to the conservative scaffolding approach that was used for the European eel genome, 20 resulting in a scaffold N50 of 17 kb. The Contig/scaffold size distribution of the common carp genome assembly is shown in Supplementary Figure S1 (supplementary material is available online at www.liebertonline.com/zeb). The genome size was predicted to be 1.4–1.5 Gbp based on k-mer counting, using an approach as described previously (Table 1). 36 Based on this assembly, we have performed gene predictions using the program Augustus. 40 Using this program, we were able to predict the presence of 82,157 genes, consisting of a total of 413,651 exons. In comparison, the zebrafish genome is predicted by Augustus to contain 53,004 genes, consisting of a total of 346,263 exons. This naïve prediction (based on gene models only) yields a considerably higher number of genes as compared to the zebrafish ENSEMBL set (based on models, empirical evidence, and manual annotations) that predicts 26,039 genes consisting of a total of 387,057 exons. At the level of individual zebrafish exons, Augustus and ENSEMBL predict approximately the same number of exons. These sets show similar amounts of homology to other genomes (see Fig. 4a). At the level of genes (complete transcripts), however, Augustus performs worse than ENSEMBL: the number of predicted genes and the level of homology deviate from ENSEMBL-based results (Fig. 4b). Analysis of individual cases suggests this to be caused by cautious behavior of Augustus in connecting exons into genes, especially when long introns need to be spanned—behavior we have also observed in our previously reported European eel genome assembly. 20 This alone would be a possible explanation for the very different number of predicted genes, but not exons, for zebrafish or carp, as the average intron length in carp is much shorter than in zebrafish. For the carp genome, the high gene (but not exon) count is not unexpected, as the assembly is still relatively fragmented, and many genes will be split amongst multiple scaffolds. The current carp genome assembly is still in the draft stage, and so are the gene predictions, and we would like to stress that they serve primarily to guide manual annotation of genes and not to give any accurate indication of the number of genes. However, it is striking that the number of predicted exons is not very much higher than in zebrafish, which is in seeming conflict with the presumed additional genome duplication of the common carp. We have performed synteny analysis and exome comparisons to investigate this further.
Comparison of carp and zebrafish gene synteny and intron size
Taking the 10 largest obtained carp scaffolds as an example, we have compared the synteny of the genes with the corresponding orthologs in zebrafish. The results for one representative carp scaffold (scaffold 1.1) is shown in Figure 2. The results for 9 other examples are shown in Supplementary Figure S2. Two points of interest can be noted: (i) The synteny of the carp and zebrafish genes is completely conserved in these 10 examples, up to the conservation of the orientation of the genes; and (ii) the total amount of noncoding sequences of the carp is on average 1.7 times smaller than that of zebrafish. This latter observation could explain why the predicted genome size of the carp is not much larger than that of zebrafish, whereas a genome duplication is expected. In order to establish whether repeat content contributes to the observed difference in the length of syntenic regions, we used Windowmasker 41 to identify repeated regions in the 10 largest carp scaffolds. Windowmasker detected 28.0% repeat content in the 10 largest scaffolds, comprising a total of 1,173,260 bp (0.1% of the assembled genome). This is significantly less than the repeat content in zebrafish (52.2% in RepeatMasker annotation of Zv9 PRJNA11776, and 52.0% in an independent windowmasking of the WGS31 zebrafish shotgun assembly CABZ00000000) and rather comparable to that of pufferfish, demonstrating the more compact organization of the carp genome. This result was further supported by an additional analysis using RepeatModeler (www.repeatmasker.org/RepeatModeler.html), leading to 11.7% masking of the same 10 scaffolds.
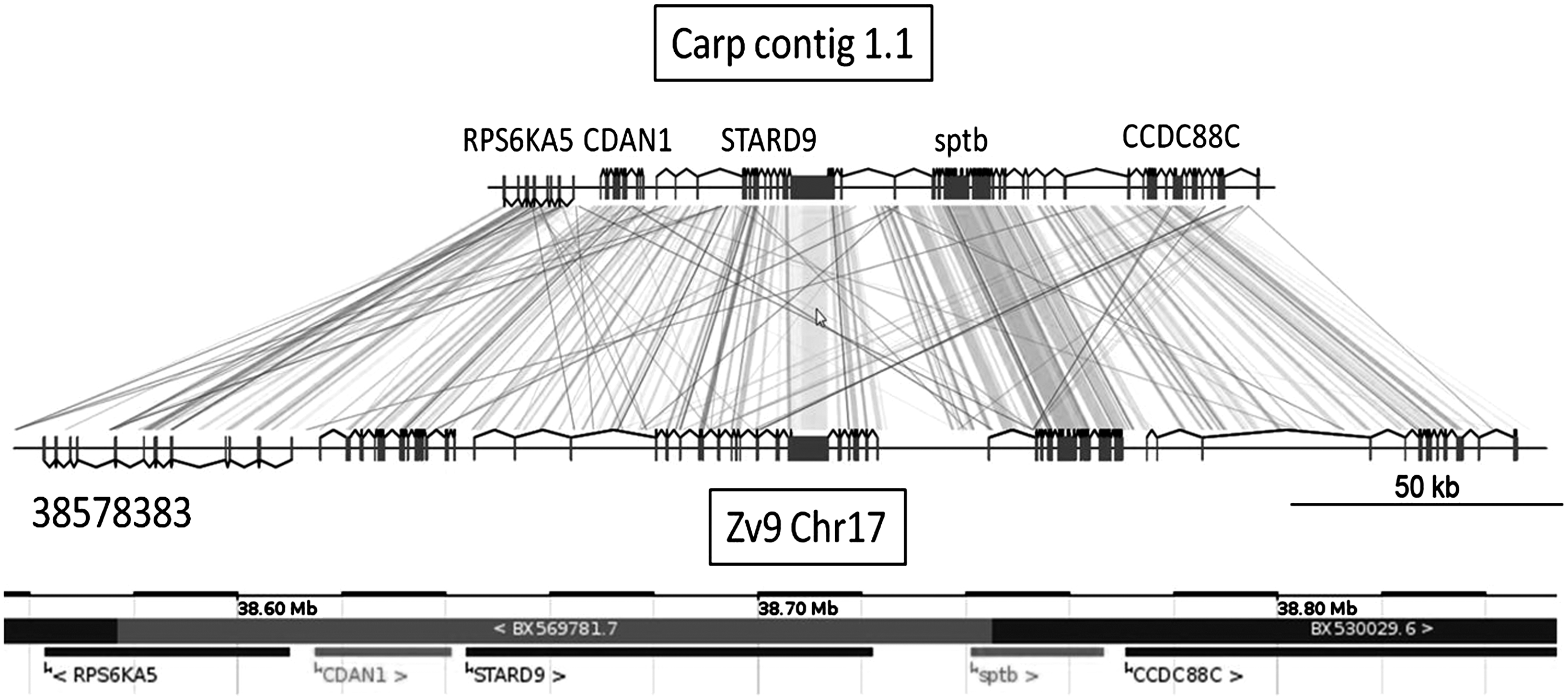
Comparison of the predicted gene structures encoded by common carp scaffold 1.1 with the corresponding homologous region of zebrafish zv9 chromosome 17 indicating Augustus gene predictions. The ENSEMBL annotation from the start of the homology region at nucleotide 38578383 of Zv9 chromosome 17 is indicated at the bottom.
A detailed analysis of the predicted gene structures shows that also at the intron level the higher compactness of the carp genome is evident: on average, the size of all predicted introns is 2.13-fold smaller in the 10 largest carp scaffolds as compared to the predicted introns in the syntenic regions in the zebrafish genome (Supplementary Table S1). We compared the Hox genes of both species since these well-known large gene clusters are good markers for genome duplication and rearrangement events.20,42 The zebrafish genome contains a total of 51 Hox genes, including 2 pseudogenes, that are divided over 7 clusters. 43 Although zebrafish has 8 Hox clusters, the HoxDb cluster does not contain any Hox genes and could only be identified based on the presence of a characteristic miRNA gene. 44 Manual annotation shows that in the common carp genome (Fig. 3), we could identify a total of 88 partial or complete Hox genes divided over 68 scaffolds. At least one complete carp orthologue was found for all, except 3 zebrafish Hox genes. No orthologues of the zebrafish pseudoHoxA10a, HoxC12b, and HoxC11b genes could be identified in common carp. For 31 out of 51 zebrafish Hox genes, including pseudoHoxA2a, we found two complete orthologues in the carp genome, and for an additional 9 out of 51 zebrafish Hox genes, we found at least part of a second orthologous gene in carp. In conclusion, only 8 out of 51 zebrafish Hox genes appear to have a single orthologue in carp, whereas the majority (40 out of 51) of zebrafish Hox genes have a partial (9 out of 51) or complete (31 out of 51) second orthologue in carp, and 3 out of 51 zebrafish Hox genes are absent in carp. In general, these data give support for an additional genome duplication event in common carp as compared to zebrafish.
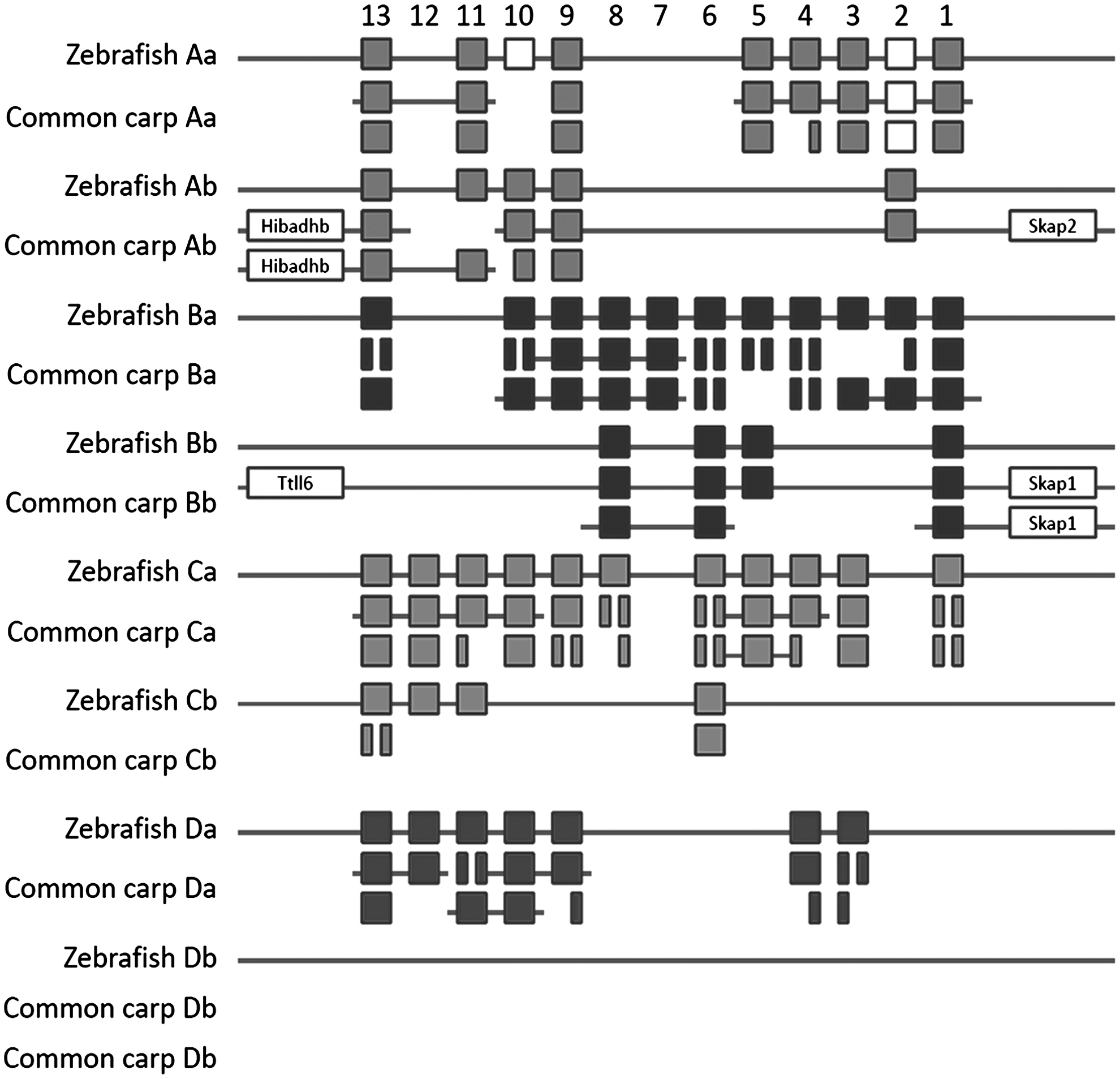
Comparison of the zebrafish and common carp hox clusters. The position of the zebrafish Hox genes is according to Corredor-Adamez et al.43 Open squares represent pseudogenes.
Comparison of the predicted exomes of common carp and zebrafish
The predicted exome of the zebrafish, as based on the ENSEMBL gene predictions was compared with the common carp genome using the BLASTN algorithm. In Figure 4a, the resulting number of hits are presented at various E-values. The genome of medaka was used for comparison. The results show that the vast majority of zebrafish exons have a very close homolog in common carp, whereas only 43 percent of exons find a hit in medaka, even at low stringency E-values. When extended to the gene level (Fig. 4b), the differences between medaka and zebrafish are less dramatic. At low stringency E-values, there remains a set of approximately 300 predicted genes in zebrafish that have no significant homologue in common carp. Of these genes, only 93 do have a representative in the Unigene or Entrez databases. GO analyses and manual annotation of this set of genes show that the majority cannot be annotated or linked to any function. In the few remaining cases (Supplementary Table S2), we find similarities with viral elements and a few genes with known function in physiology or development, such as several chemokines (Supplementary Table S2). In a reverse comparison, the exome of the common carp was predicted using the software program Augustus and the resulting set of predicted transcripts was blasted against the zebrafish. In this analysis, transcripts were chosen over exons in order to avoid that the complexity of splice variants would make comparisons extremely difficult to interpret. Furthermore, the specific examples described below were checked manually at the exon level. In the initial BLASTN result, 7952 predicted transcripts did not give a hit on the zebrafish genome (Table 3). This set of sequences was further compared with the zebrafish genome using TBLASTX. The results show that there remain 1238 sequences without a significant homolog. A comparison with transcriptome data sets that we obtained from embryos and adults of the same subspecies of common carp 30 (see also Material and Methods section) shows that a majority of 698 of the predicted transcripts are expressed in at least embryos or adult carp. This indicates that these predictions represent common carp genes that are lacking in the zebrafish genome. Manual BLAST analyses were performed with a subset of 47 predicted transcripts of larger than 500 nucleotides that could not be identified at all in the zebrafish genome at any stringency (Table 3). The results (Supplementary Table S3) show that there are 17 expressed predicted transcripts that have clear homologs in other species than zebrafish. Most related are homologs in other fish species. The presence of a putative TLR gene and 2 putative lymphocyte markers again indicate subtle differences in the immune systems of carp and zebrafish. This approach of unbiased BLAST-based data mining using the differences between closely related species therefore seems a good approach to identify interesting new genes involved in fish-specific functions. Furthermore, the transcriptome data confirms that the majority of all transcripts are covered at least partially by the genomic assembly (Table 3 and data not shown).
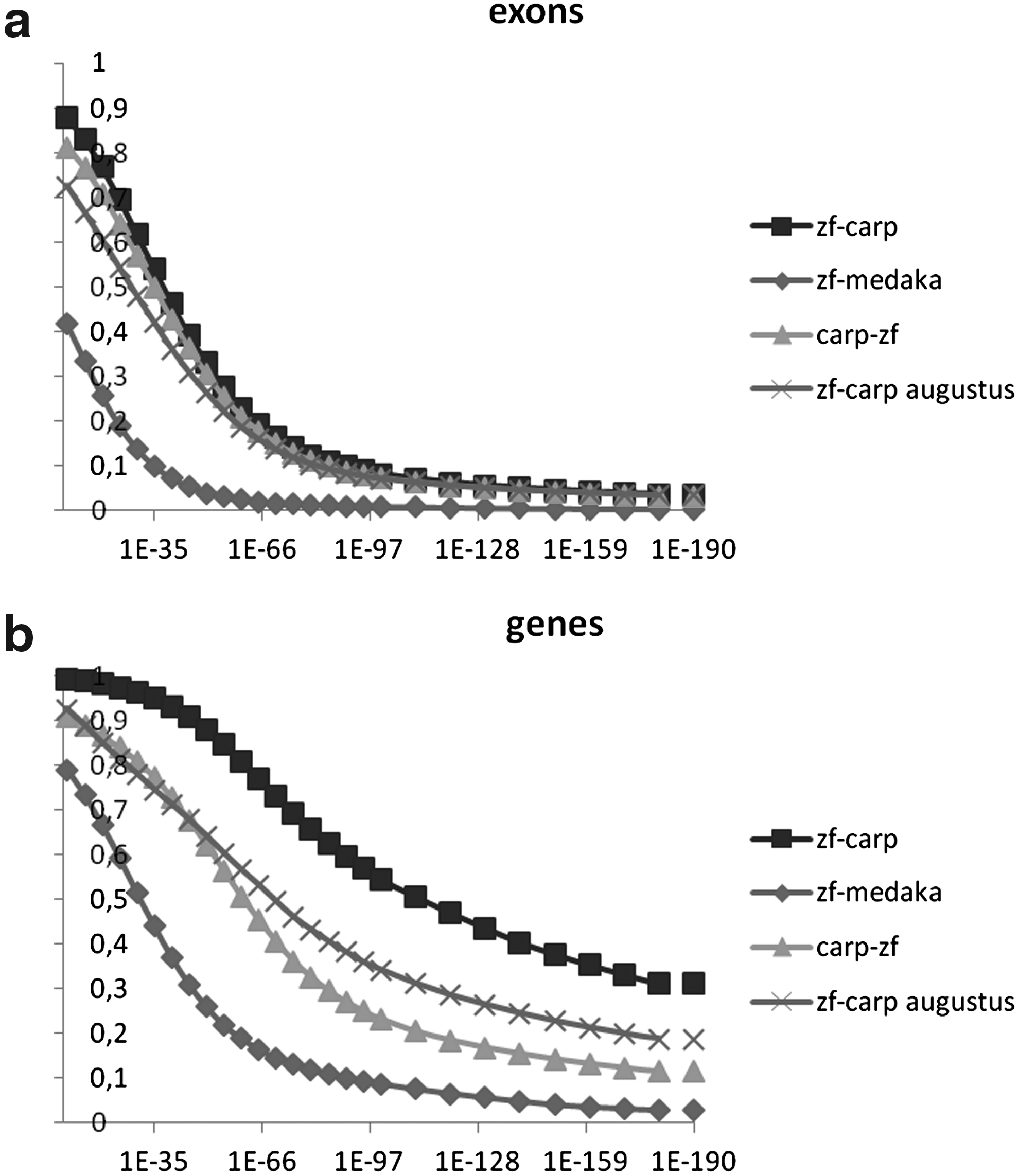
BLAST comparisons of the exomes of zebrafish, common carp, and medaka. In all cases, carp comparisons were based on predictions based on the program Augustus; (
Expression in RNAseq data was analysis using RPKM analysis of the predicted transcripts in four RNA deep sequencing sets.
See Supplementary Table S3 for detailed manual annotation of blast hits.
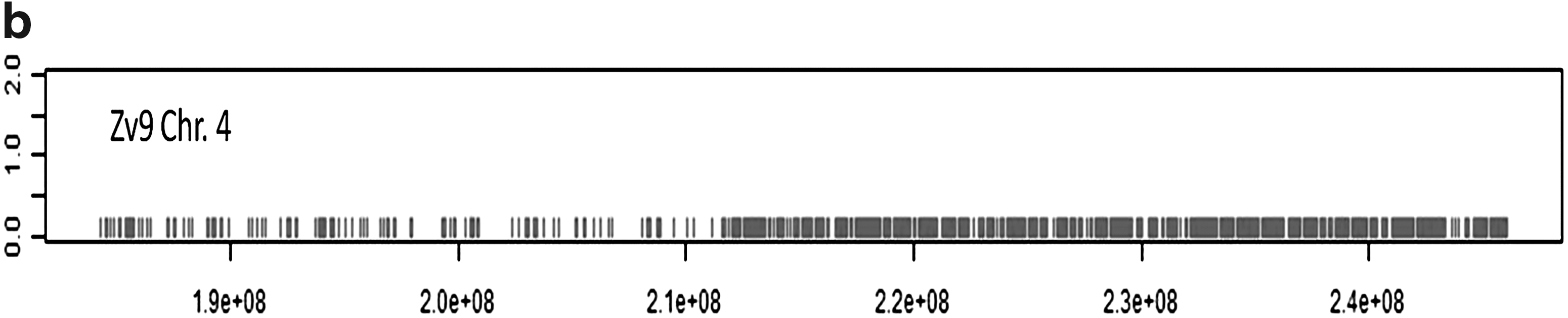
The results shown in Figure 5 demonstrate that a large number of exons of zebrafish have two hits in the carp genome. Reversely, only a small minority of predicted exons of common carp have two hits in the zebrafish genome. Surprisingly, there are some zebrafish exons that have more than 250 highly similar copies in the carp genome (Fig. 5b), whereas the reverse is not observed. Analysis of the 250 exons indicates that many of them belong to genes of transcription factors or genes containing NACHT domain patterns. In addition, there are a number of predicted carp exons that give over 4000 hits in the zebrafish genome. As seen in Figure 6, these hits are not randomly distributed over the zebrafish genome but have a remarkable enrichment in the right q half of chromosome 4 (Fig. 6b). As a control, we performed analysis of zebrafish predicted exons on the zebrafish genome. This analysis shows also a highly unequal distribution of highly duplicated predicted genes on chromosome 4 and chromosome 22, very similar to the figures shown in Figure 6 (data not shown). These parts of the zebrafish genome are characterized by highly duplicated miRNAs and other repeat areas and a sparse distribution of confirmed gene sequences. This part of the zebrafish genome is quite exceptional and will be described in detail elsewhere (K. Howe, manuscript in preparation).
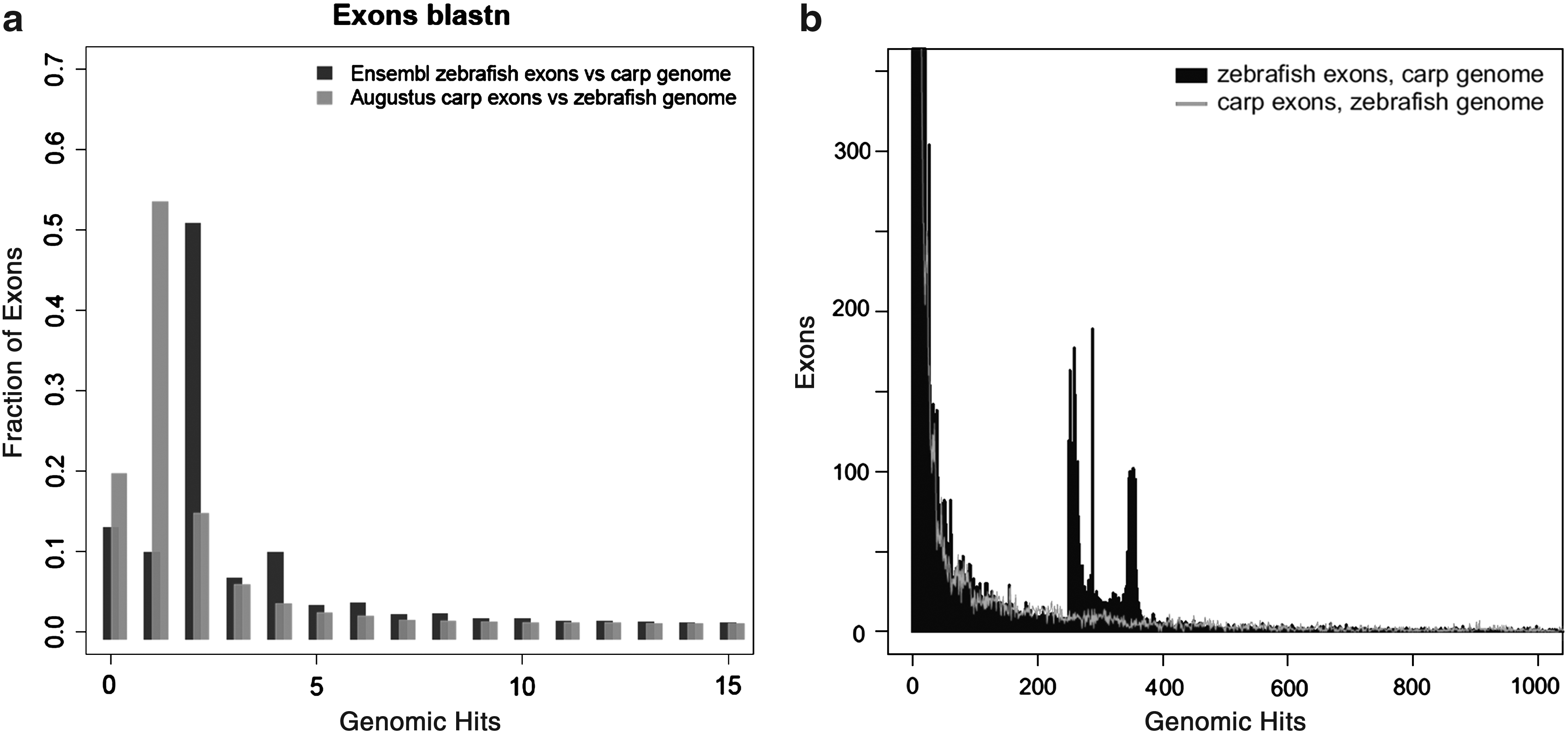
Quantitation of the multiplicity of BLAST hits. (

Chromosomal location of carp sequences with multiple blast hits in the zebrafish genome. (
Discussion
The common carp, Cyprinus carpio, is a very close relative of the model species zebrafish, Danio rerio. Research on common carp can be of benefit for zebrafish research because of resources available owing to its large body size, such as the availability of sufficient organ material for transcriptomics, proteomics, and metabolomics. Vice versa, functional genomic studies in zebrafish can be of benefit for research on common carp, and therefore lead to new applications in aquaculture, especially since at present cyprinid fish are the most cultured fish for food production worldwide. From an evolutionary point of view, the comparison of two closely related cyprinid species from different climate zones with very different body mass and spawning characteristics will be of great interest, especially considering the availability of highly inbred clonal carp lines. Comparative genomic studies will give insight into events such as additional genome duplication events that occurred in the carp lineage.
In this study, we give a concise description of a draft genome assembly of the European subspecies of common carp (Cyprinus carpio carpio). This species is predicted to have undergone a genome duplication event as compared to other relatives of the Cyprinid fish family such as zebrafish, and this is confirmed by the observed extended duplication of its predicted gene repertoire as shown in this article. As indicated by detailed analysis of the Hox clusters, we can conclude that there has been moderate gene loss in the originally hypothesized genome duplication. As compared to the zebrafish genome, we detected fewer repetitive sequences. This apparent compactness allows for a similar size of both genomes despite the carp genome indicating pseudo-tetraploidity in the absence of extensive loss of duplicated genetic material. The very high level of microsynteny between the carp and zebrafish genomes makes these organisms highly suitable for further functional genomic comparisons. Data-mining based on comparative BLAST analyses showed interesting differences in various unique gene sets, of note for our research being the presence of species-specific immune genes in both species. For future functional studies in zebrafish, our data present a valuable additional resource for comparisons allowing studies that require large amounts of tissue material as possible in common carp. Reversely, with the availability of the common carp exome, genetic information obtained from the zebrafish model can now more efficiently be used for future studies in common carp, which owing to its importance for aquaculture can also offer industrial applications. With the increased availability of common carp transcriptome datasets based on RNA deep sequencing, such as presented here or recently described,30,45,46 the genomic data will lead to reliable gene predictions for the entire genome that hopefully soon will reach a same level as that currently already available for zebrafish. Our data will hopefully stimulate follow-up bioinformatic endeavors to further integrate common carp gene predictions with the available zebrafish gene databases, for example at ENSEMBL and ZFIN. Furthermore, our preliminary analysis of the genome such as statistical analysis of repeat sequences, has given a basis for future evolutionary analyses. The obtained genomic data set for the common carp represents an important resource for functional and genomic follow up studies that will make use of the high similarities between carp and zebrafish. The high coverage of the genomic sequence ensures that most if not all common carp genes have at least obtained partial sequence coverage. Last but not least, the quality of the common carp draft genome was shown to be sufficient to identify all expected gene homologs from zebrafish and thereby presents a major resource for future gene data mining in this organism. The lack of polymorphic alleles in our clonal homozygous carp line made the obtained sequence data suitable for data mining approaches based on BLAST searches, demonstrating its usefulness for future genetic research. We plan to combine this resource in future research with extensive transcriptome data sets, for instance, for a multitude of organs and tissues obtained from fish under healthy and disease conditions.
Footnotes
Acknowledgments
We thank Dr. F. Verbeek and E. Stupka for stimulating discussions and the company Baseclear (Leiden The Netherlands) for hosting the Illumina sequencing equipment and fruitful collaborations. The authors gratefully acknowledge the financial support of the Smart Mix Program (![]() ) of The Netherlands Ministry of Economic Affairs and of The Netherlands Ministry of Education, Culture and Science awarded to Leiden University and to ZF-screens B.V.
) of The Netherlands Ministry of Economic Affairs and of The Netherlands Ministry of Education, Culture and Science awarded to Leiden University and to ZF-screens B.V.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.