
Abstract
Significance:
Generalized lymphatic anomaly and Gorham–Stout disease are extremely rare diseases with severe symptoms and poor prognosis. The etiology and clinical presentation of the patients remain poorly defined, but recent research has attempted to determine the pathogenesis of these diseases.
Recent Advances:
In recent years, the characteristics of complex lymphatic anomalies have been revealed. Kaposiform lymphangiomatosis is recognized as a new entity that has an aggressive course and poor prognosis. Genetic analysis revealed somatic mutations in genes associated with the phosphoinositide 3-kinase (PI3K) pathway in lymphatic malformation lesions. Somatic NRAS mutation in lymphatic endothelium from a generalized lymphatic anomaly patient was also detected as a potential cause of disease. Furthermore, studies demonstrated the efficacy of the mammalian target of rapamycin (mTOR) inhibitor sirolimus for these lymphatic diseases.
Critical Issues:
These diseases have overlapping symptoms, imaging features, and complications, leading to difficulty in their differential diagnosis. In addition, there are no standard therapies. Therefore, we need to determine the differences among these diseases to not only diagnose but also treat them appropriately.
Future Directions:
Further investigations should reveal differences in the clinical features and findings of radiological, pathological, and genetic examinations to manage each disease appropriately. Somatic mutation in genes encoding RAS/PI3K/mTOR signaling pathway components could be associated with the pathogenesis of these diseases and may be novel targets for drug therapies.
Michio Ozeki, MD, PhD

Scope and Significance
Generalized lymphatic anomaly 1 –3 (GLA), Gorham–Stout disease 4,5 (GSD), Kaposiform lymphangiomatosis 6 –8 (KLA), and channel-type lymphatic malformations (LMs) are classified according to their pathology 1 (Table 1); however, some disease forms with differing pathologies are not separately defined entities. 9 Furthermore, central conducting lymphatic anomaly (CCLA), a channel-type LM 1,2 with unclear etiology, might represent a spectrum of lymphatic pathologies. Because clinical findings overlap, and disease etiology, management, outcomes, and sequelae are unclear, intractable LMs (termed complex lymphatic anomalies [CLAs]) 2 are difficult to diagnose. We review recent studies reporting different clinical characteristics of vascular anomalies to further understand their pathogenesis and optimal treatment.
International society for the study of vascular anomalies classification of vascular anomalies
The causative gene.
CCLA, central conducting lymphatic anomaly; GLA, generalized lymphatic anomaly; GSD, Gorham–Stout disease; KLA, kaposiform lymphangiomatosis; LMs, lymphatic malformations; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha.
Translational Relevance
Congenital structural abnormalities of lymphatic vessels and the thoracic duct, or abnormal proliferation of local lymphatic endothelial cells may cause the abnormal conditions in these patients. Vascular endothelial growth factor (VEGF)-C can induce controlled lymphatic recovery after injury. 10 Thus, VEGF-C pathological models are important for understanding the pathogenesis of CLAs. 11 –13 Recent studies have investigated lymphangiogenesis and lymphatic vasculature remodeling and suggested that genetic abnormalities in the RAS/mitogen-activated protein (MAPK) and phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) signaling pathways could be responsible for these lymphatic diseases. 14 –18
Clinical Relevance
CLAs involve multiple organs and have diverse symptoms, but an appropriate diagnosis is difficult as their clinical findings overlap (Fig. 1). Notably, osteolytic lesions are useful for distinguishing between GLA/KLA and GSD. 19 Patients with KLA have a severe coagulation disorder and life-threatening hemorrhagic pericardial and pleural effusion. 20 The histological hallmark of KLA is kaposiform, hemosiderotic, spindle-shaped lymphatic endothelial cells arranged in clusters. 6 Intranodal lymphangiography and dynamic contrast-enhanced magnetic resonance lymphangiography (DCMRL) are new imaging techniques able to detect the anatomical findings of the thoracic duct and lymphatic flow, which can lead to a correct diagnosis of CCLA. 21
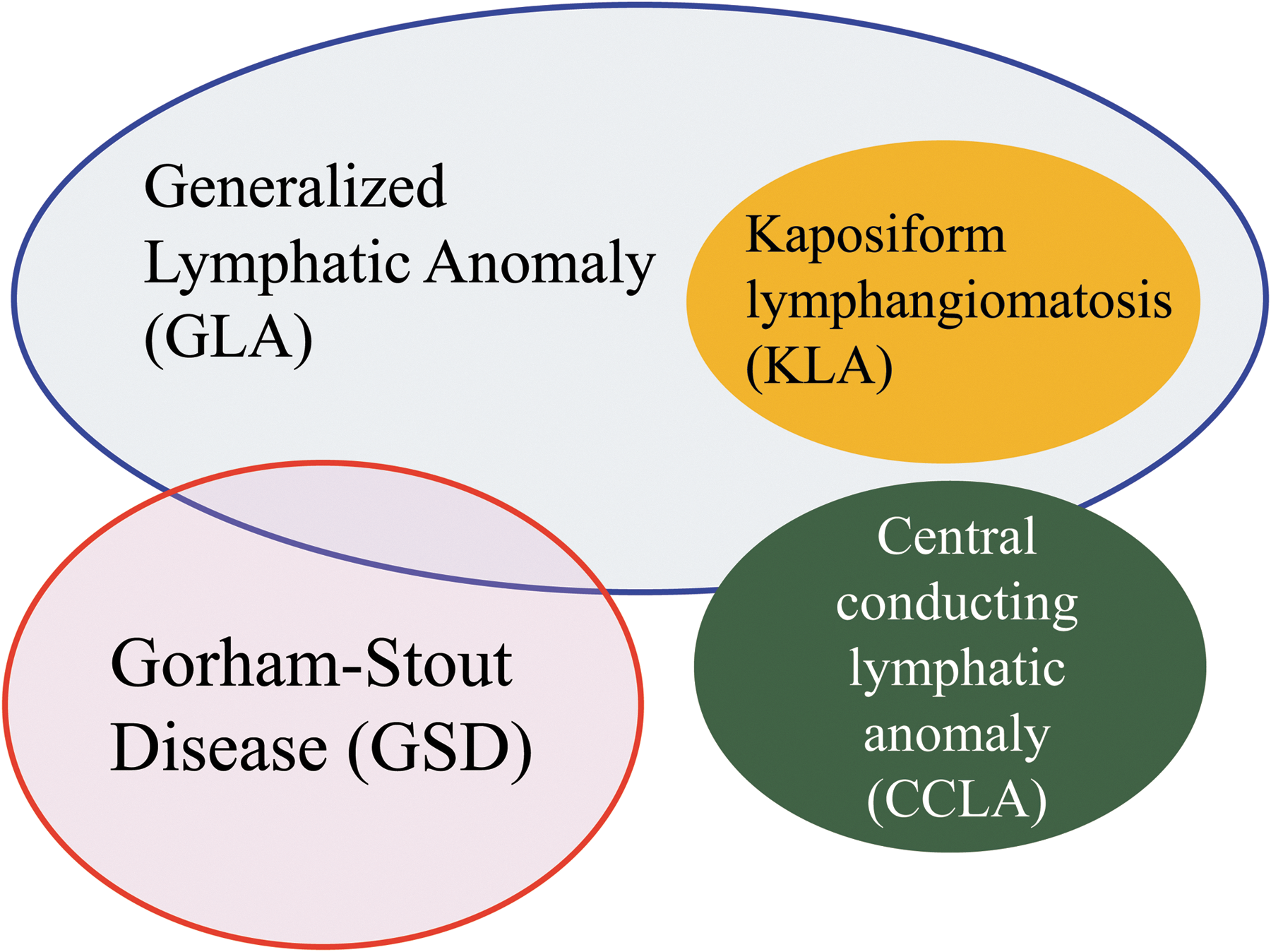
Overlapping features of complex lymphatic anomalies. Complex lymphatic anomalies are overlapping their clinical symptoms and characteristics.
Discussion of Findings and Relevant Literature
LMs in International society for the study of vascular anomalies classification
In 1996, the first International Society for the Study of Vascular Anomalies (ISSVA) classification was approved at the 11th workshop. In 2014, an updated classification was approved at the 20th workshop. This classification has been promoted to spread correct understanding of vascular anomalies. When clinicians and scientists plan their clinical and basic research, they often confuse inconsistent terminology. The correction and simplification of that terminology and systematization of guidelines have promoted a more uniform approach to researching vascular anomalies. Common (cystic) LMs (macrocystic, microcystic, or mixed) are the most common lymphatic anomalies that occur in infants. These lesions are solitary and cystic-appearing soft-tissue masses that commonly arise in neck, mediastinum, and retroperitoneum. Their typical appearance and symptoms help us to differentiate them from CLAs. The updated classification categorized GLA and GSD as separated systemic LMs. 22 GLA is a rare multiorgan disease involving organs such as lung, mediastinum, spleen, and proliferating lymphatic vessels diffusely. Its thoracic involvement can cause respiratory failure and is associated with poor prognosis. This condition predominantly affects children and adolescence and has variable symptoms. GSD is a rare and enigmatic disease characterized by the presence of an intraosseous LM. The osteolysis progresses destructively with the developing and proliferating LM. In the World Health Organization classification, KLA was distinguished as a rare tumor of lymphatic vessel origin. 23 However, KLA was defined as a provisionally unclassified vascular anomaly in the updated ISSVA classification of 2014 because KLA patients have invasive and aggressive features, but the pathological findings show a hypoproliferative rate. KLA is described as an aggressive disease of the lymphatic system and has foci of “kaposiform” abnormal spindle lymphatic endothelial cells. 6 –8 In a review from 2000, Faul described how some patients with a GLA called diffuse pulmonary lymphangiomatosis showed the presence of spindle-shaped cells around endothelial cells, which contrasted with primary pulmonary lymphangiectasia, 9 but this was not designated as a specific disease entity. Both GLA and KLA patients have multiple organs affected and the lesions can cause similar symptoms such as pleural effusion, ascites, and skin involvements. In the 2018 updated ISSVA classification, KLA was categorized as a subtype of GLA 1 (Table 1). In contrast, channel-type LM, CCLA, previously called lymphangiectasia, is characterized by dilated lymphatic channels, dyskinesia, or obstructive impairment. The abnormalities of lymphatic structure can induce problem of clearance of lymph. 2 The symptoms depend on the site of the anomaly, such as primary intestinal lymphangiectasia, pulmonary lymphangiectasia, which causes protein-losing enteropathy, pleural effusion, peripheral lymphedema, and lymphorrhea. Primary lymphedema is sporadic, or rarely familial lymphatic malformative disease. It mainly involves the lower extremities and the lymphatic subcutaneous fibroadipose lesions are not curable and grow gradually. It has been determined that most types of primary lymphedema are caused by various genetic mutations. 1 Against this background, in some patients, it is difficult to achieve a correct diagnosis of CLAs because the clinical findings overlap among these conditions. There has been a lack of information on not only their etiology but also their specific management, long-term outcomes, and disease sequelae. However, recent studies on the spectrum of CLAs have suggested that there are significant differences in their clinical characteristics. Further study should be performed, leading to a more comprehensive understanding of the etiology and optimal treatment for CLAs.
Etiology and genetics
The causative genes of LM
The development of next-generation sequencing has identified low-level somatic mutations in sporadic lesions of vascular anomalies, 15 and the updated ISSVA classification now contains a list of causal genes. 1 There are over 40 genes that are associated with the etiology of vascular anomalies. Although primary lymphedemas are caused by germline mutation, LMs are sporadic, not familial, and caused by somatic changes in components of the PI3K/AKT/mTOR pathway (Fig. 2). Mutations in PIK3CA have been found in 16 of 17 specimens of cystic LM (mutant allele frequency, 0.8% to 10%). 24 Five PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha) mutations (p.E542K, p.H1047R, p.C420R, p.E545K, and p.H1047L) were detected in patients with LM. However, patients with other vascular malformative/overgrowth disorders also had the same mutations. Another report investigated isolated lymphatic endothelial cells from a patient who had the angiogenic phenotype. The authors identified two mutations in these lymphatic endothelial cells that showed higher proliferation and AKT activation than those of human lymphatic endothelial cells. 17 These features can form part of a syndrome such as congenital lipomatous overgrowth, vascular malformations, epidermal nevi, scoliosis/skeletal and spinal (CLOVES) syndrome, Proteus syndrome, and Klippel–Trenaunay syndrome, which feature lymphatic disruption and overgrowth. Recent studies have shown that PIK3CA-related overgrowth spectrum (PROS) is caused by somatic mosaicism of variants in genes of the PI3K pathway. 25 PROS includes hemihyperplasia multiple lipomatosis, CLOVES syndrome, macrodactyly, fibroadipose hyperplasia or overgrowth, Klippel–Trenaunay syndrome, megalencephaly–capillary malformation (MCAP or M-CM), fibroadipose infiltrating lipomatosis/facial infiltrative lipomatosis, and dysplastic megalencephaly. 1 Although the terms used to describe vascular anomalies have been made more scientific by the ISSVA based on histopathological findings, differentiation of these diseases is challenging based on their phenotypic presentation alone because patients within this spectrum of overgrowth syndromes have overlapping clinical features.
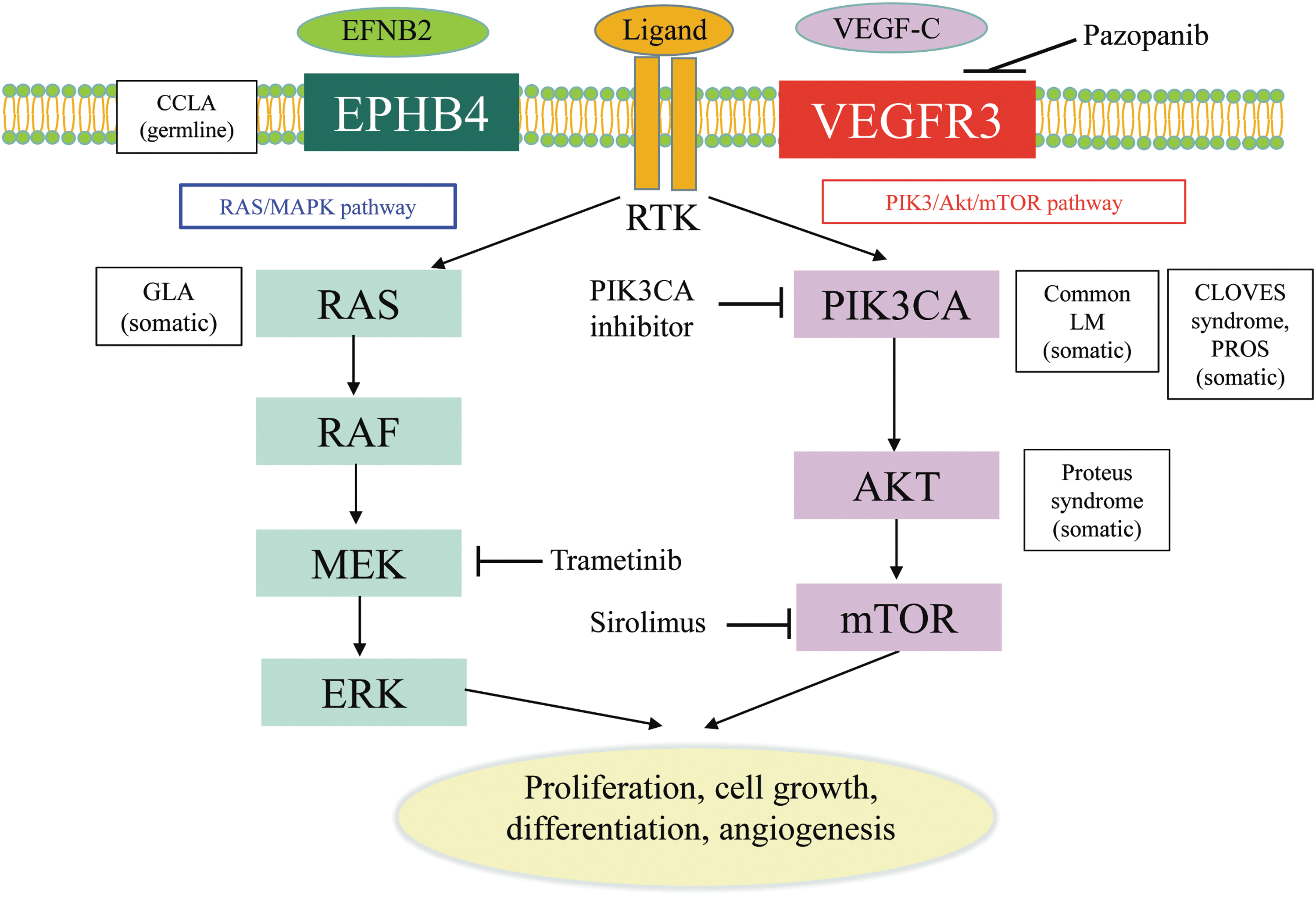
Mutations and signaling pathways involved in LMs. Mutations in EPHB4 and NRAS lead to activate the signaling of RAS/MEK/ERK pathway. Mutations in PIK3CA and AKT lead to activate the signaling of PIK3/AKT/mTOR pathway. CLOVES, congenital lipomatous overgrowth, vascular malformations, epidermal nevi, scoliosis/skeletal and spinal; EFNB2, ephrin-B2; EPHB4, ephrin B4; GLA, generalized lymphatic anomaly; LM, lymphatic malformation; mTOR, mammalian target of rapamycin; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PROS, PIK3CA-related overgrowth spectrum; RTK, receptor tyrosine kinase; VEGF-C, vascular endothelial growth factor-C; VEGFR, VEGF receptor.
Regarding other CLAs, little has been reported on the associated genetic abnormalities. However, Manevitz-Mendelson et al. reported the possibility that somatic NRAS mutation causes GLA. 18 A variety of human malignancies have activating mutations in RAS proto-oncogenes (KRAS, HRAS, and NRAS), which have an important role in carcinogenesis. 26 Manevitz-Mendelson et al. analyzed lymphangiomatosis endothelial cells (LyECs), which were isolated from a GLA patient using CD31-coated magnetic beads. A somatic mutation in NRAS gene (c.182A >G, Q61R) in fewer than 30% of the alleles was identified in LyECs. 18 In addition, the NRAS mutation plays key roles in the regulation of angiogenesis and lymphangiogenesis. Treatment with an mTOR inhibitor, sirolimus, and an MEK inhibitor, trametinib, had an effect of reducing the viability of the LyECs through inhibition of the phosphorylation of AKT and ERK, and so might be a novel target treatment of GLA. Furthermore, another group found that CCLA might be associated with a germline mutation in EPHB4. 27 Ephrin B4 (EPHB4) encodes ephrin B-type receptor 4 that binds to ephrin B2 (EFNB2). The EFNB2/EPHB4 pathway plays a role in the determination of venous and lymphatic cell fate and lymphatic valve development. 28,29 In this study, the zebrafish model of EPHB4 mutation was shown to mimic the lymphatic presentation of CCLA, including the abnormality of lymphatic vessel branching and formation. The model demonstrated that this mutation might be responsible for the differentiation defects of lymphatic vessels in CCLA patients. This can also be effectively reduced by treatment with sirolimus and trametinib. These studies demonstrated that these genes can cause the pathogenic etiology of these diseases and the inhibition of these genes might be a target for treatment.
The mechanisms of osteolysis in GSD
The mechanisms behind the osteolysis in GSD are unknown, but various hypotheses have been proposed. It is known that osteolytic lesion in GSD is associated with local proliferation of lymphatic vessels. 30 The activation of osteoclasts and lymphangiogenesis are essential for the development of GSD and are repeatedly observed in its lesions (Figs. 3 and 4). Osteolysis is also seen in GLA and KLA, but these osteolytic lesions consist of diffuse dilated lymphatic endothelial cells. Therefore, the etiology of GLA and KLA is distinct from that of GSD. Although, lymphatic vessels are abnormally distributed in affected bone lesions in GSD, and normal bone shows an absence of such vessels. The mechanism and triggers of invasion of lymphatic endothelial cells into bone remain unknown. It is considered that activation of the osteoclastogenic process is induced by immune mechanisms in affected bone tissue because the number of osteoclasts increases in the GSD lesion. 31 Alternatively, osteolysis could be caused by the abnormal proliferation of lymphatic vessels compressing bone. Moreover, the activity of osteoclasts is influenced by some cytokines that are secreted by the lymphatic endothelial cells. The endothelial cells express VEGF receptors (VEGFRs) and VEGF signaling plays an important role in angiogenesis and lymphangiogenesis. 32 The receptor for VEGF-C/D, VEGFR-3, is a key regulator of lymphangiogenesis. 33 A high local concentration of VEGF-C or other prolymphangiogenic factors (VEGF-A, VEGF-D, and angiopoietin) can induce angiogenesis and lymphangiogenesis in the bone marrow of patients with GSD. In fact, VEGF-C level is increased in the serum of patients. 34 A recent study also revealed that the transgenic overexpression of VEGF-C in bone stimulates the formation of bone lymphatics and osteoclast-mediated bone resorption that resembles GSD. 13 Circulating levels of C-terminal telopeptide of type I collagen (CTX-1), a small peptide generated by osteoclast-mediated cleavage of collagen I, 35 were found to be significantly higher and the number and activity of osteoclasts were increased in VEGF-C transgenic mice. Furthermore, osteal macrophages and activated T cells secrete various cytokines and promote angiogenesis and lymphangiomatosis (Fig. 3). Activated T cells produce the ligand for receptor activator of nuclear factor κB (RANK) and osteoprotegerin (OPG) binds to ligand for RANK (RANKL), which inhibit interaction with RANK and osteoclastic differentiation and activation. 36 Type 2 T helper cells, osteoblasts, and dendritic cells produce interleukin (IL)-6 and can promote the angiogenesis and differentiation of osteoclasts. IL-6 increases bone resorption through the RANKL induction of mesenchymal cells. The recent study reported by Hominick et al. 13 revealed a crucial point, but it is unclear why the local level of VEGF-C increases and triggers abnormal angiogenesis and lymphangiogenesis at GSD lesions. An important issue in this context is what induces the immunological triggering of osteal macrophage and endothelial cell activation. This might be associated with somatic gene mutation or inopportune function of bone-resident cells. To clarify this, further investigations should be performed using affected tissue samples. The main factors involved in this triggering may be an appropriate and effective target for the drug treatment of GSD.
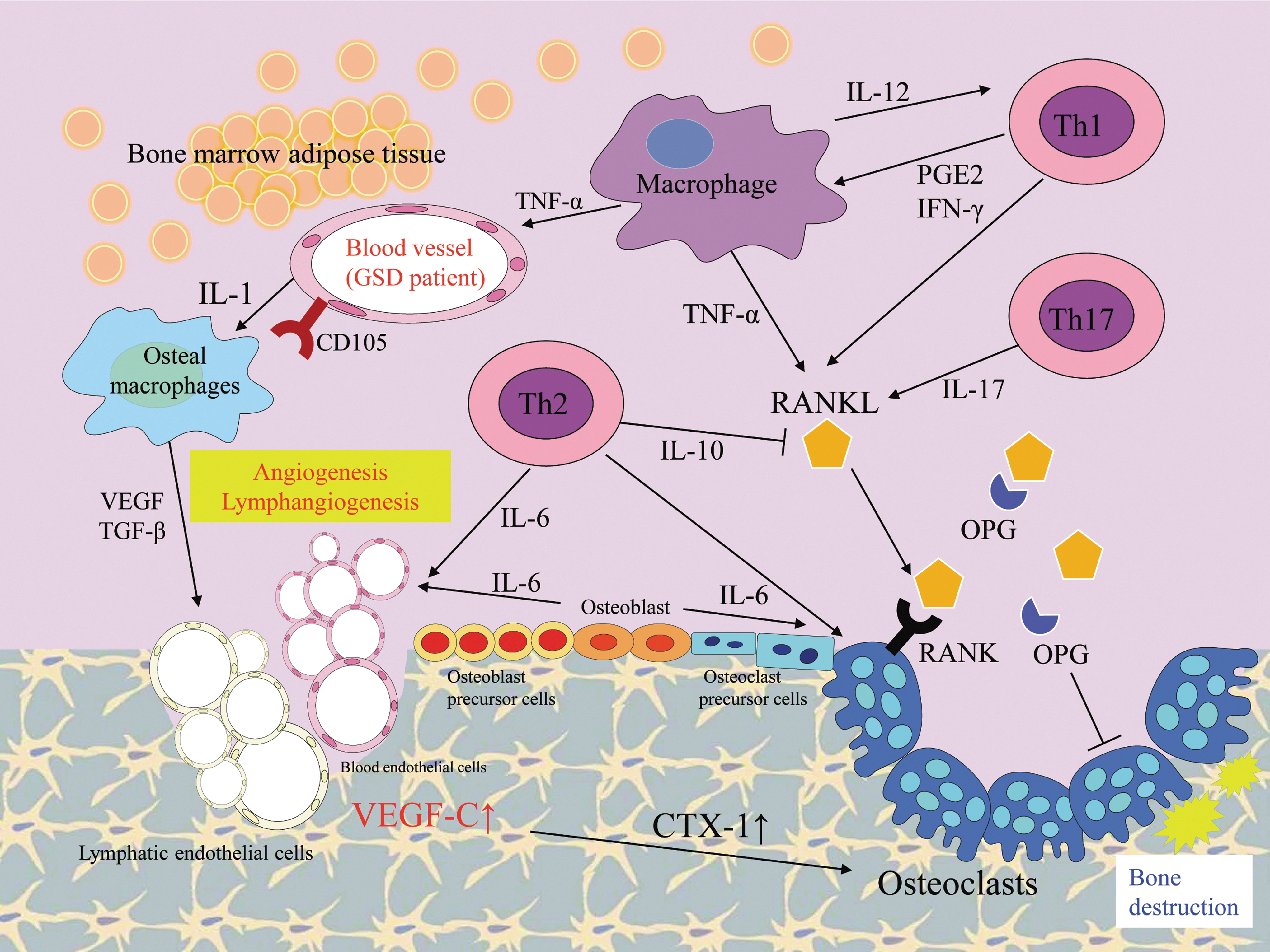
Schematic diagram summarizing the possible pathway of key elements in GSD. In this schema, immunological responses, related secreted cytokines, and cells are shown. The arrows indicate the activated directions and lateral lines indicate suppressive actions. The development of lesions in GSD has been associated with angiogenic and lymphangiogenic factors. The formation of bone lymphatics and osteoclast-mediated bone resorption can be seen in affected bone lesions of GSD patients. The endothelial cells in GSD patients highly express CD105 and activate osteal macrophages by producing IL-1. VEGF and TGF-β produced by osteal macrophages promote angiogenesis and lymphangiomatosis. CTX-1, C-terminal telopeptide of type I collagen; GSD, Gorham–Stout disease; IFN, interferon; IL, interleukin; OPG, osteoprotegerin; PGE2, prostaglandin E2; RANK, receptor activator of nuclear factor κB; RANKL, ligand for receptor activator of nuclear factor κB; TGF, transforming growth factor; Th, helper T; TNF, tumor necrosis factor.
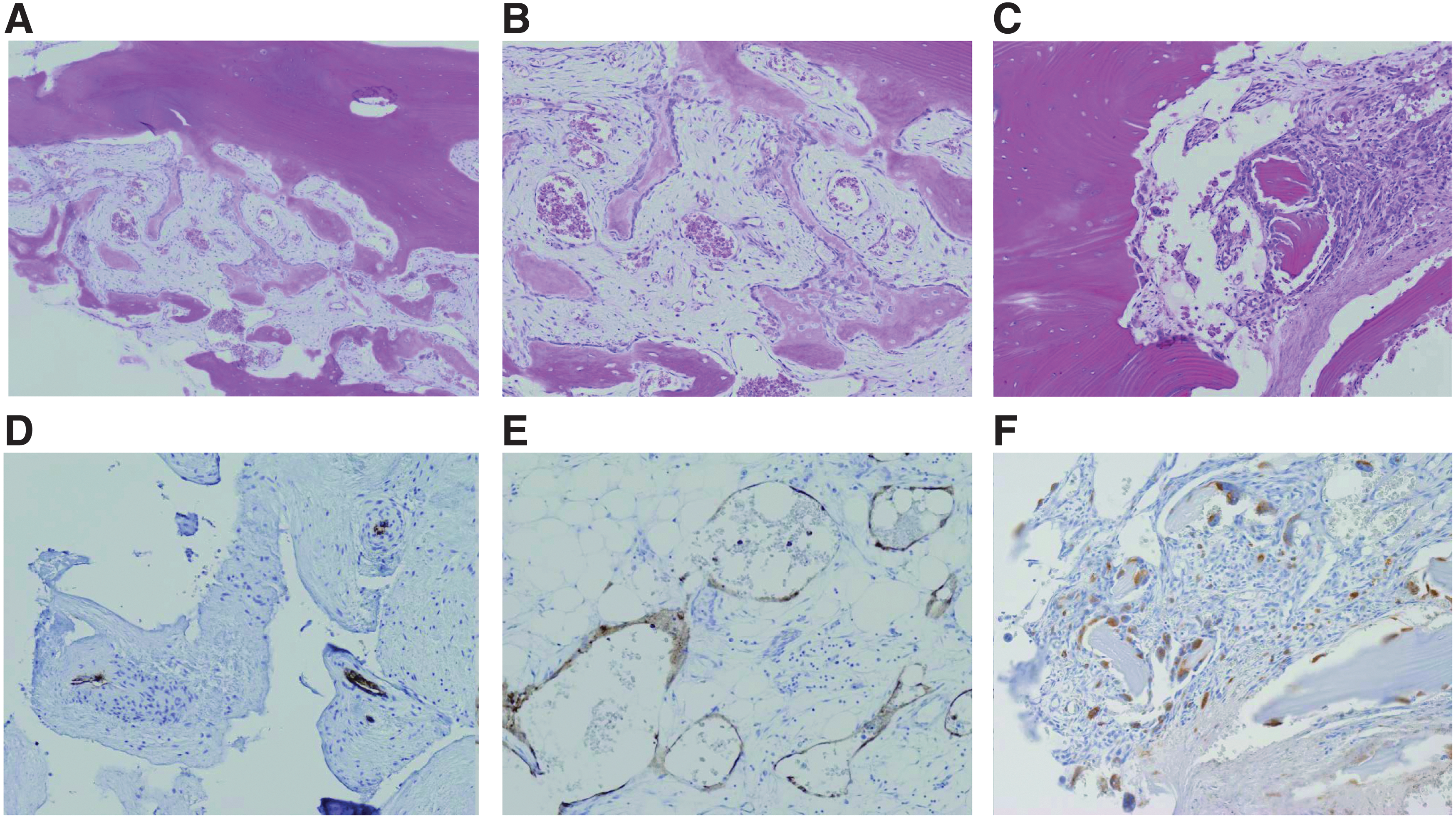
Pathological findings in patients with GSD. In the pathological examinations of GSD patients, diffuse lymphatic vessel proliferation with progressive osteolysis and the activation of osteoclasts are the characteristic features.
Clinical features and differential diagnosis of CLAs
The characteristics of CLAs and differential features from other diseases are shown in Table 2. CLAs involve multiple organs, have diverse symptoms, and are common in children and adolescents. One report described how LM proliferation during childhood is different from that in adults and that these LMs proliferate at a significant rate. 37 The common characteristics of these diseases are lymphatic tissue lesions, including osteolytic lesions, thoracic lesions (chylothorax, pleural effusion, pericardial effusion, mediastinal mass, and retroperitoneal soft-tissue mass), abdominal lesions (ascites, splenic lesions, and retroperitoneal mass), and skin lesions (lymphedema and lymphorrhea). However, to achieve a precise diagnosis, a variety of assessments, clinical, imaging, histological, and hematological examinations, are needed. 2,38 In terms of the clinical laboratory findings, CLA patients often have hypoalbuminemia, hypoproteinemia, hypogammaglobulinemia, anemia, and lymphocytopenia caused by lymph leakage (chylothorax, pleural effusion, and ascites). They also have moderate or mild thrombocytopenia, low fibrinogen, and coagulopathy (increasing D-dimer and fibrinogen degradation products), called chronic localized intravascular coagulopathy (LIC). 1,2,39 In general, vascular malformations with slow flow are associated with risk of LIC and deep vein thrombosis. The mechanisms of coagulopathy in CLAs are unknown, but are considered that platelets are involved by abnormal lymphatic endothelium entrapping. The Kasabach–Merritt phenomenon (KMP), which leads to more severe coagulopathy and thrombocytopenia, is associated with vascular tumors, tufted angioma, and kaposiform hemangioendothelioma (KHE). KLA patients also have more severe LIC and thrombocytopenia (50,000–100,000/μL) than those with LM and GLA. A recent study also showed that the serum angiopoietin 2 level in KLA and KHE with KMP patients was higher than in controls. 40 Therefore, KLA and KHE with KMP may have similar phenotypes and pathogeneses associated with the presence of abnormal spindle cells.
Characteristics of complex lymphatic anomalies and their differential features
Ang, angiopoietin; BAP, bone-specific alkaline phosphatase; CTX-1, C-terminal telopeptide of type I collagen; IL, interleukin; OPG, osteoprotegerin; PIL, primary intestinal lymphangiectasia; RANKL, ligand for receptor activator of nuclear factor κB.
There is a need to rule out other osteolytic diseases in the differential diagnosis of GSD. Idiopathic multicentric osteolysis, multicentric osteolysis, hereditary multicentric osteolysis, neurogenic osteolysis, and osteolysis due to neoplastic, infectious, and immunological diseases should be considered before reaching a final diagnosis. A series of laboratory investigations were carried out, but they failed to detect osteolysis secondary to pathology (cyst, tumor, and infection), metabolic and endocrinal disorders, neoplastic disease, trauma, and/or connective tissue disorders. A recent case report showed the elevation of bone-specific alkaline phosphatase (BAP) and serum CTX-1 in a GSD patient; the patient showed rapidly decreasing levels of BAP and CTX-1 after bisphosphonate therapy. 41 Another report showed the elevation of serum tartrate-resistant acid phosphatase before bisphosphonate therapy. 42 At initial assessment, the elevation of serum IL-6 was found, but the level of IL-6 normalized after 6 months of bisphosphonate treatment. In contrast, the levels of the osteoclastic regulators OPG, free soluble RANKL, and OPG/free RANKL ratio were increased. In GLA and KLA patients, serum angiopoietin 1 and 2 levels were found to be useful for their diagnosis and suitable as biomarkers for drug treatment. 40 In terms of new radiological methods, Papadakis reported that 18 F sodium fluoride positron emission tomography/computed tomography (CT), which is used for detecting skeletal metastatic disease, is useful for GSD diagnosis and follow-up. 43 However, the findings obtained by these methods are only speculative, preventing definitive conclusions from being drawn.
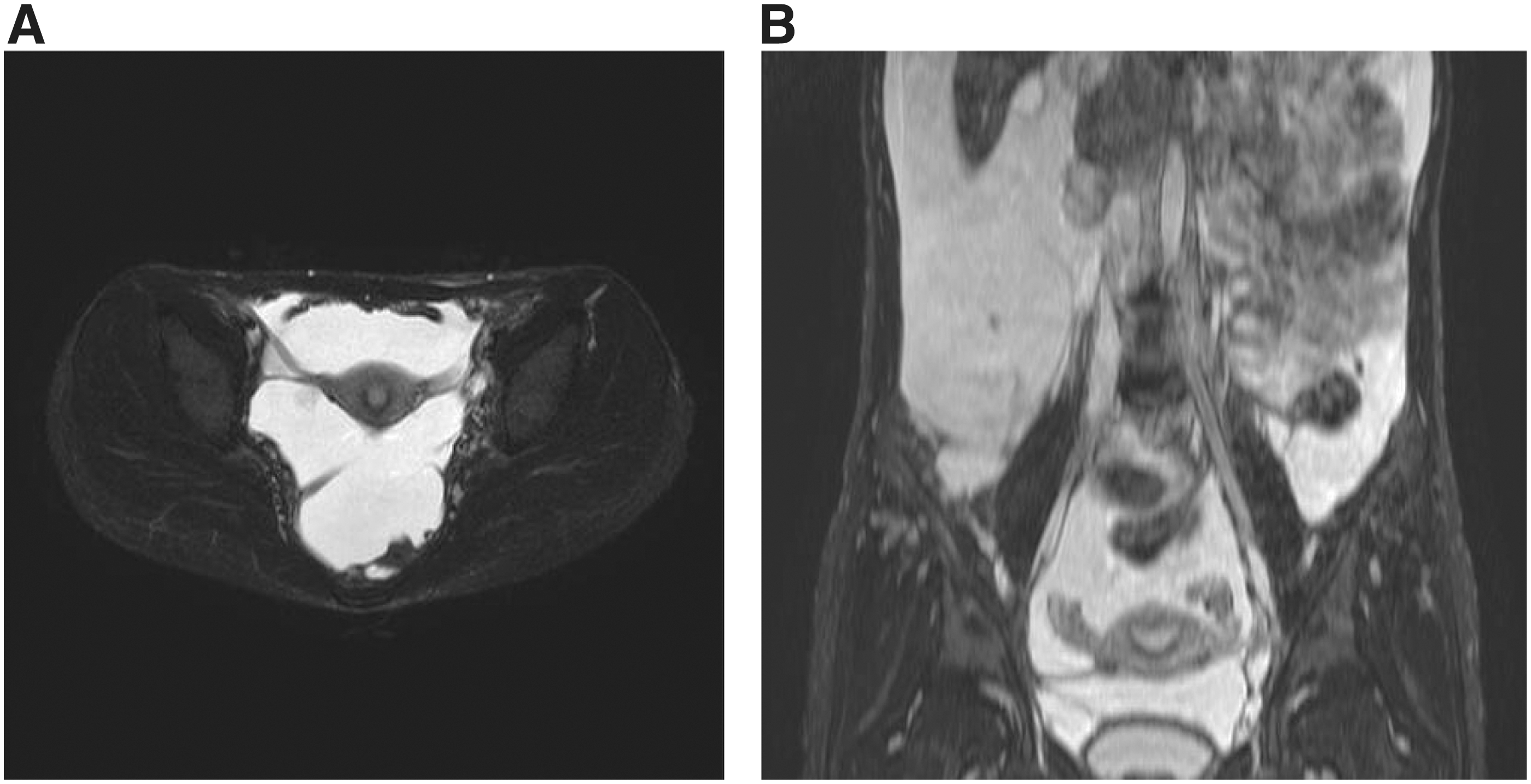
Although there are some overlapping features, the radiological findings in the osteolytic lesion of GSD patients are different from those of GLA/KLA patients. In patients with GLA/KLA, the medullary cavity of osteolytic area shows lytic areas. In contrast, GSD lesions are characterized by progressive osteolysis and the loss of cortical bone 19 (Figs. 5 and 6), which can extend beyond adjacent joints. The number of affected bone lesions in GLA/KLA is also typically higher compared with GSD. The osteolytic lesion of GSD is commonly localized to the cranium, ribs, clavicle, and cervical spine. Destructive osteolytic lesions can cause pathological fractures, pain, and swelling. However, small multiple lytic lesions might not cause pain in GLA patients (Fig. 6). The GSD patient often has progressive and infiltrative osteolytic lesions (Fig. 5); in contrast, osteolytic lesions of patients with GLA/KLA are not progressive. Furthermore, GLA and KLA patients have vertebral involvement more frequently than GSD patients. 44 The lumbar spine is the most commonly affected site in GLA and KLA patients.
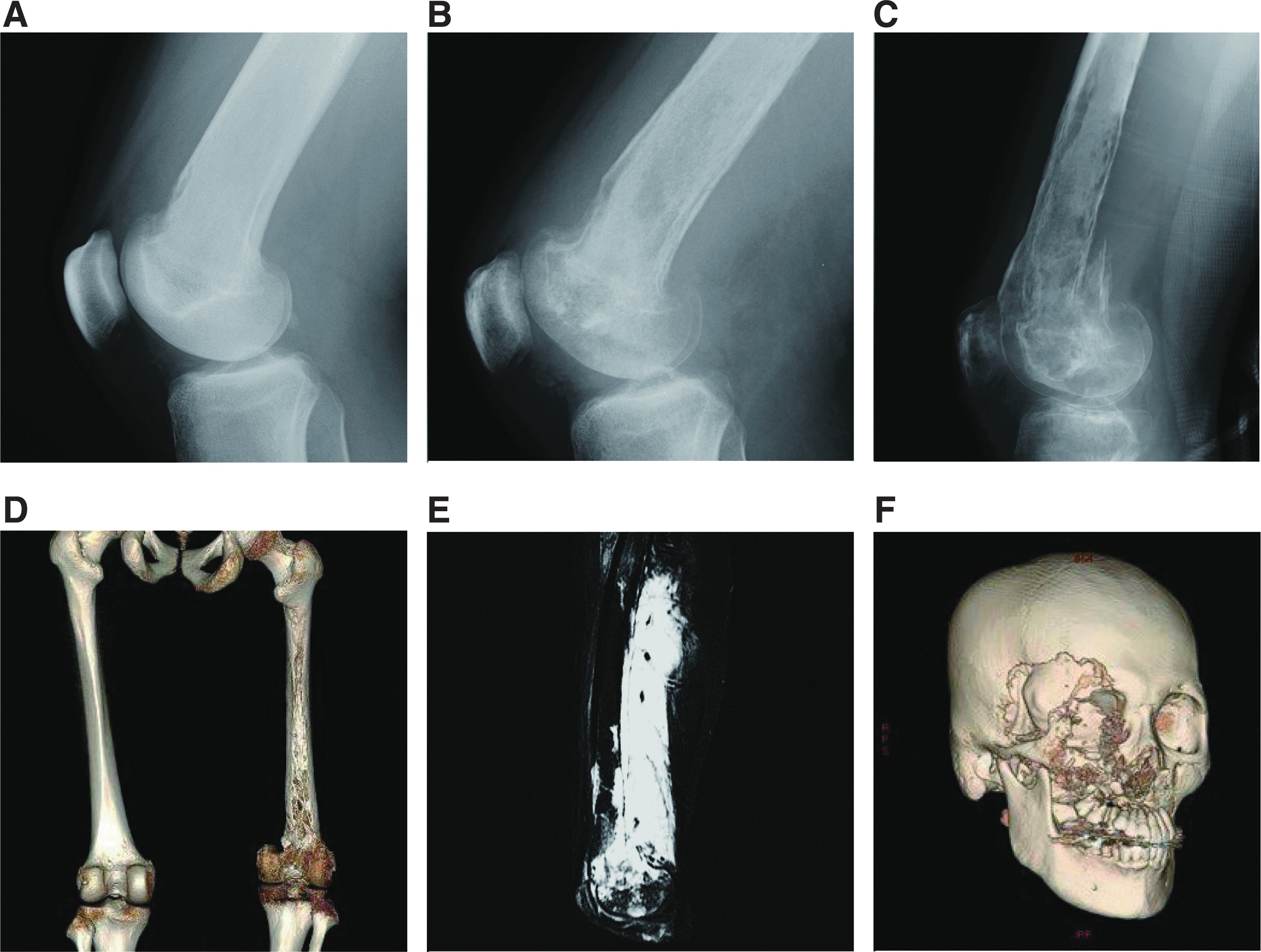
Radiological findings of patients with GSD. The osteolytic imaging of GSD patients is characterized by progressive, destructive cortical resorption that can extend beyond adjacent joints.
Radiological findings of patients with GLA and KLA. The multiple lytic areas in the medullary cavity are shown in the osteolytic images of GLA and KLA.
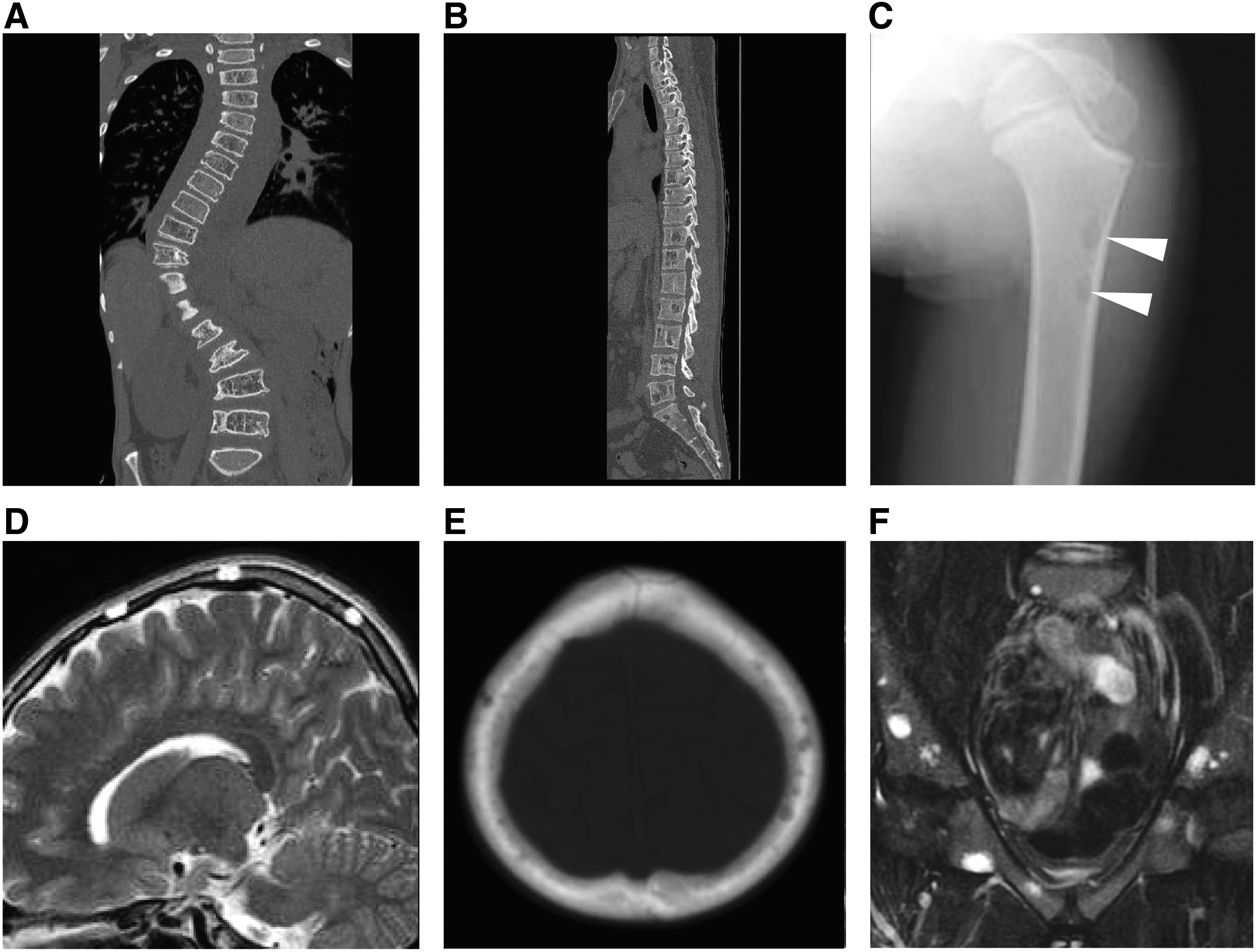
In our previous article, we reported the characteristics of GLA, KLA, and GSD. 20 The following symptoms were more common in GLA/KLA than in GSD: pleural effusion, mediastinal mass, and cardiac effusion. Pleural and cardiac effusion in KLA were similar to that in GLA; KLA patients more frequently have hemorrhagic pericardial and pleural effusions and mediastinal masses than GLA patients. A deteriorating clinical course, retroperitoneal infiltrative soft-tissue thickening, hemorrhagic effusions,, and associated thrombocytopenia are also characteristic of KLA patients. 45 The mortality in KLA is significantly poorer than GLA (mortality rates: KLA, 55.6%, 5/9; GLA, 28.6%, 10/35, p = 0.0268). 20 Mortality was associated with progressive pulmonary disorders and serious coagulation disorder. The involvement of shoulder girdle or thoracic vertebral bone is predominantly associated with pleural effusion. KLA patient has thickening of bronchovascular bundles and interlobular septa more than GLA 46 (Fig. 7).
Radiological images of visceral lesions in KLA patients. KLA patients often have mediastinal masses and hemorrhagic pericardial and pleural effusion. In radiological examination, thickening of bronchovascular bundles, retroperitoneal infiltrative soft-tissue thickening, and interlobular septa are common findings.
If the clinician cannot diagnose the disease by radiological examinations, biopsy of the lesion is the next diagnostic step. Both GLA and KLA show dilated malformed lymphatic channels, but KLA partially shows foci of patternless clusters of spindle cells 20 (Fig. 8). However, it is not necessary to biopsy all GLAs to rule out KLA. The key features of KLA are thrombocytopenia, coagulation disorder, and hemorrhagic pericardial and pleural effusion or ascites. The presence of an atypical or aggressive pattern can be used to rule out lymphangiosarcoma or other diseases. KHE also features spindle cells and abnormal lymphatic channels, and we need to distinguish KHE or KLA based on the clinical symptoms and distribution of the lesions. 47 Biopsies of bone tissue specimens are small and often crushed; so poor samples may lead to misdiagnosis. Some articles recommend not performing a rib biopsy because of the high risk of the subsequent development of refractory pleural effusions. 48 If the lesion spreads superficially, a biopsy of the surface should be performed, although it may not be easy to stop lymph leakage. Idiopathic chylothorax and chylous ascites, which are hard to treat, occur without any invasive examination or treatment. Therefore, if such leakage into the closed lumen is observed, the biopsy can be performed from the lesion on the surface of the thoracic or abdominal cavity.
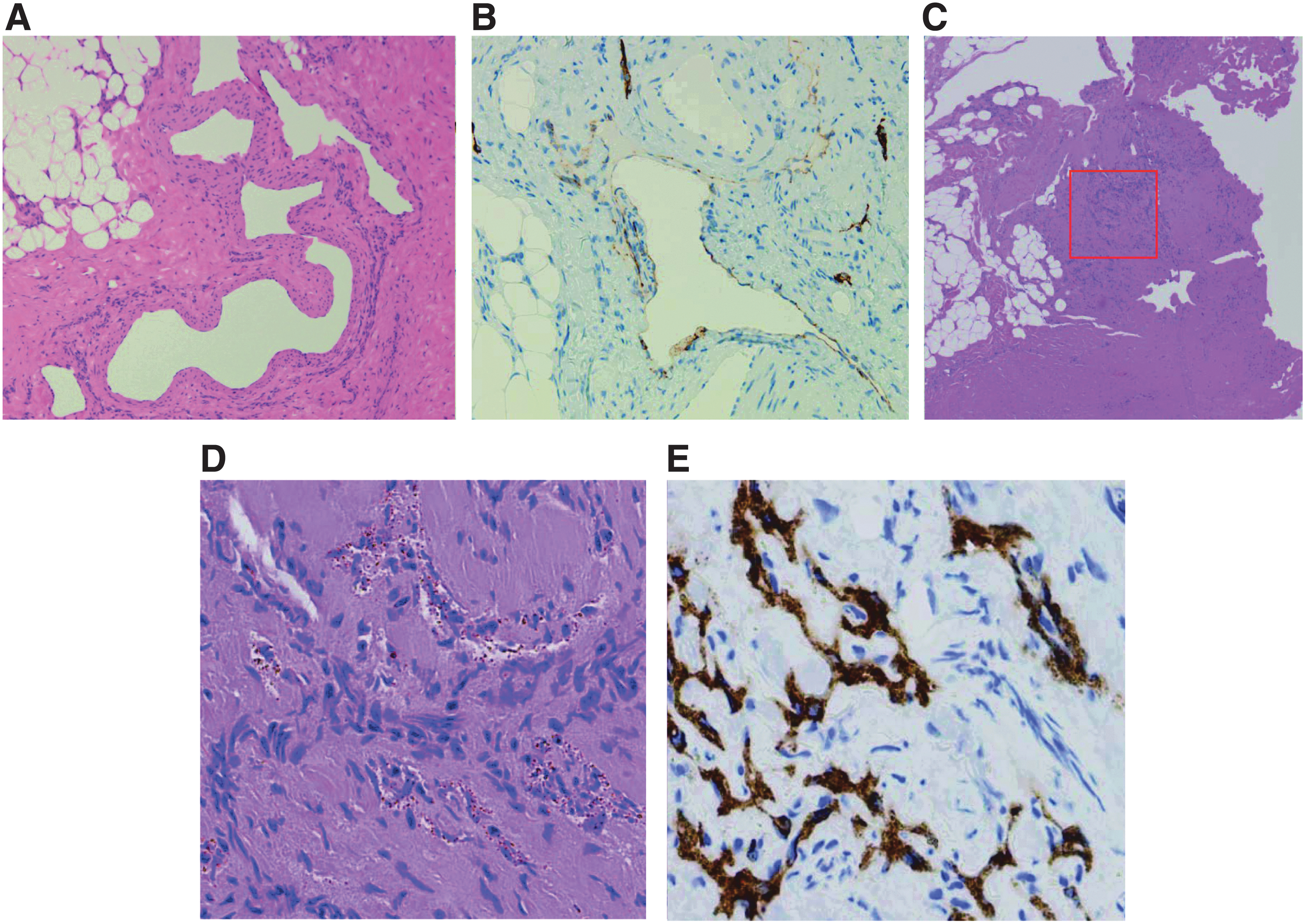
Pathological findings in patients with KLA. In GLA and KLA patients, the typical pathological examinations show dilated malformed lymphatic channels However, foci of patternless clusters of lymphatic spindle cells are characteristic of the patients with KLA. This is associated with, platelet microthrombi, and some degree of fibrosis.
Patient assessment, monitoring, and follow-up
Because CLA patients have variable symptoms and problems, they should be consulted by a multidisciplinary team of specialists. Physical assessments and monitoring are very important for overall assessment, monitoring, and follow-up. However, standardized assessment methods for patients with lymphatic anomalies have not yet been established. In general, the following assessment procedures are used: clinical symptoms, radiological evaluation, the amount of effusion, and quality-of-life scale. The clinician should consider the methods used and frequency of examination by condition of the patient and affected lesion(s). If symptoms suggestive of bone lesions are present, patients should undergo screening of the whole body, involving both a skeletal survey and a bone scan. In addition, patients should be evaluated using not only plain radiography but also ultrasound, CT, and magnetic resonance imaging (MRI) to detect adjacent soft-tissue lesions. If no bone lesions are detected, frequent monitoring is not required. GSD patients have progressive bone resorption, so we recommend monitoring by plain radiography of the target bone lesion at least once every 2 months. In contrast, GLA and KLA patients have slowly progressive bone lesions, so we recommend monitoring by plain radiography of the target bone lesion annually. We should also conduct a skeletal survey, as the bone lesion may spread to other parts. Fat-saturated T2-weighted MRI can detect minimal lymphatic lesions (for example, skull bone lesions in GLA patients) that are difficult to detect on plain radiography.
Patients are evaluated using imaging such as ultrasound, CT, and MRI for visceral or adjacent soft-tissue lesions. Lymphoscintigraphy is rapid and minimally invasive without major complications compared with lymphangiography. However, lymphangiography is superior in being able to show detailed images, such as detecting the exact site of leakage of the chylothorax. 2,48 Regarding the diagnosis of thoracic CLAs, such imaging at an early stage is recommended to be performed to detect the leakage-causing lesion because it can help to obtain not only a precise diagnosis but is also useful information for treatment selection. 49 A new method, DCMRL, was shown to distinguish central conducting lymphatic dilation from normal tissue of retroperitoneal, which can give us helpful information to diagnose CCLA. 50,51 The ligation or occlusion of dilated central conducting lymphatics may be an effective therapy for the patients with intractable pleural effusions or ascites.
Dysfunction of musculoskeletal system and wounds
CLAs involve disorders of the musculoskeletal system and skin, and patients with CLAs sometimes have trouble with wounds. GSD patients often exhibit osteolytic lesions of the appendicular skeleton and have infiltrative soft-tissue lesions surrounding the osteolytic lesion. Soft-tissue lesions show high signal intensity at fat-saturated T2-weighted MRI and demonstrate intense enhancement following the administration of gadolinium contrast agent. These lesions can lead to chronic pain, lymphedema, lymphorrhea, joint abnormalities, asymmetric girth, leg length discrepancy, and scoliosis. Chronic lymphedema or lymphorrhea leads to refractory bleeding, chronic infection, and dermal hypertrophy. Cases with severe cutaneous involvement can exhibit chronic ulceration and angiosarcoma (Stewart–Treves syndrome). 52 In addition, 35% of KLA patients have subcutaneous masses that are soft, are not tender or painful, and do not grow. 8 These patients undergo resection, but care should be taken regarding the possibility of local lymphorrhea in the resected area. In such cases, it can sometimes be difficult to stop the leakage. These patients should undergo consultations with specialists such as orthopedists, dermatologists, and plastic surgeons in combination as early as possible.
Treatment
Clinicians should treat CLA patients corresponding to their condition, complications, and the size and location of the lesion(s). Surgery and interventional therapy are performed in patients with lymphatic anomalies. However, drug treatments are limited and prospective clinical trials are lacking. It is difficult to cure extensive or complicated lesions using only local therapy, so multimodal therapy is necessary. We should begin with conservative treatments (for example, nutrition therapy) if the condition of the patient is not life threatening. The administration of albumin/immunoglobulin and blood transfusion may also be required depending on the patient's symptoms. Aggressive treatment, including medical and surgical treatment, is necessary for cases of more than moderate severity. Bisphosphonates that inhibit bone resorption are used for the treatment of patients with osteolysis. 53 Meanwhile, interferon, which inhibits the proliferation of blood and lymphatic vessels, is used for patients with bone or generalized lymphatic lesions. 54 Other pharmaceuticals include the anti-VEGF-A antibody bevacizumab, 55 propranolol, 56 steroids, vitamin D, and calcitonin. Although these drugs, in single or combinatorial use, can improve conditions of some patients, effectiveness is limited.
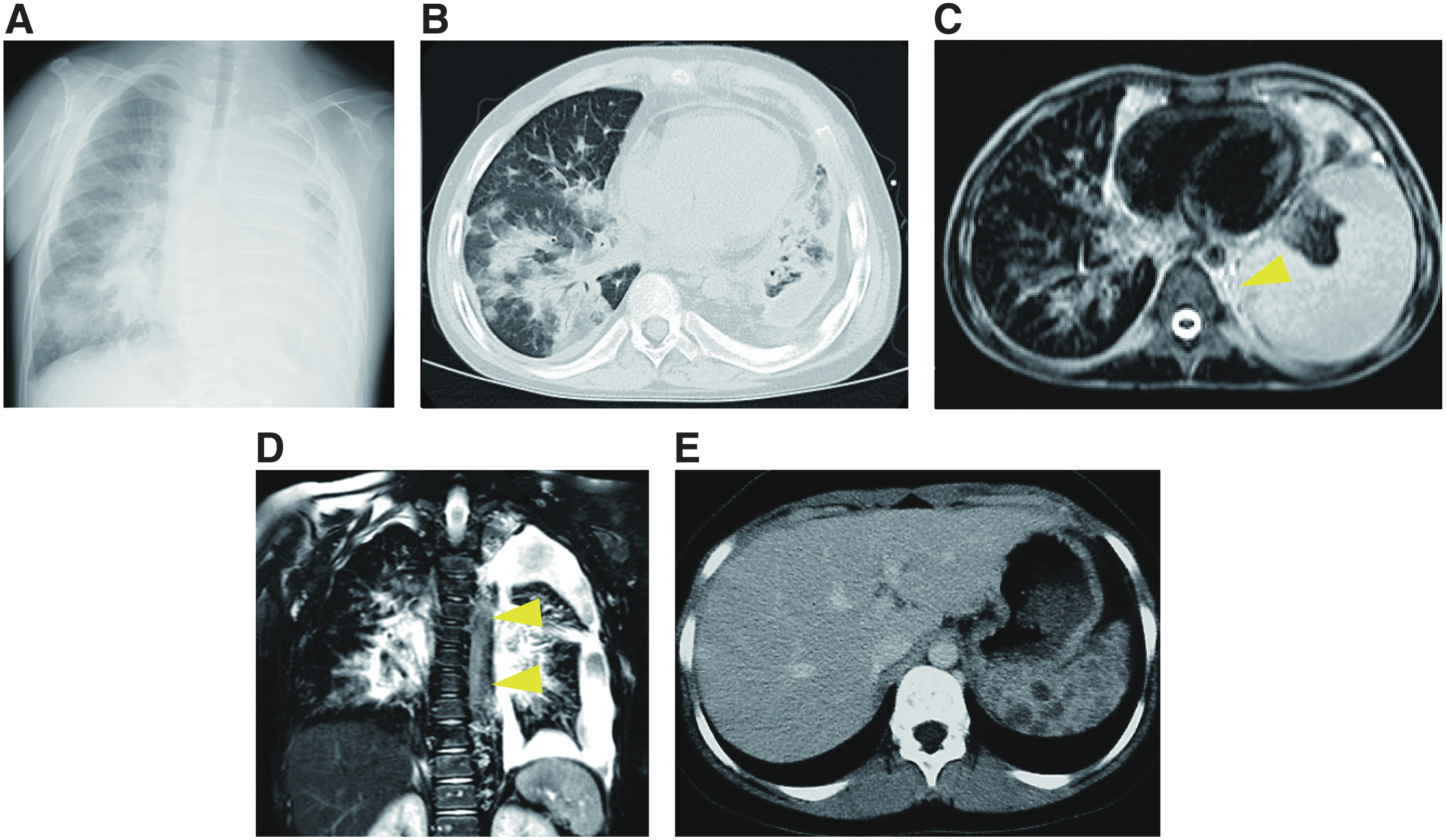
In recent studies, the role of the PIK3/AKT/mTOR pathway has been found in the development of blood vessels and lymphatic tissues, and new therapeutic agents that target this pathway are under development. The mTOR inhibitor sirolimus is known to inhibit lymphangiogenesis, and is thought to act on lymphatic tissues within lesions to regulate the production and leakage of lymph by decreasing lymphatic endothelial cell activity 57 –60 (Figs. 9 and 10). In lymphangiectasia model mice, Baluk et al. demonstrated that sirolimus inhibited the growth of abnormal lymphatics and reduced lesions without adverse effects on normal lymphatics. 61 In these treated lymphatic endothelial cells, the expression of Prox1 and VEGFR-3 decreased without the caspase-dependent apoptosis. A prospective trial by Adams et al. also indicated that sirolimus had an excellent effect on vascular anomalies, with a good response observed in CLA patients. 60 This trial reported three distinct assessments involving radiological examination, functional impairment score, and quality of life of the patients. Notably, 100% (7/7) of patients with GLA, 100% (3/3) of patients with GSD, and 71% (5/7) of patients with KLA showed a partial response at 6 months.
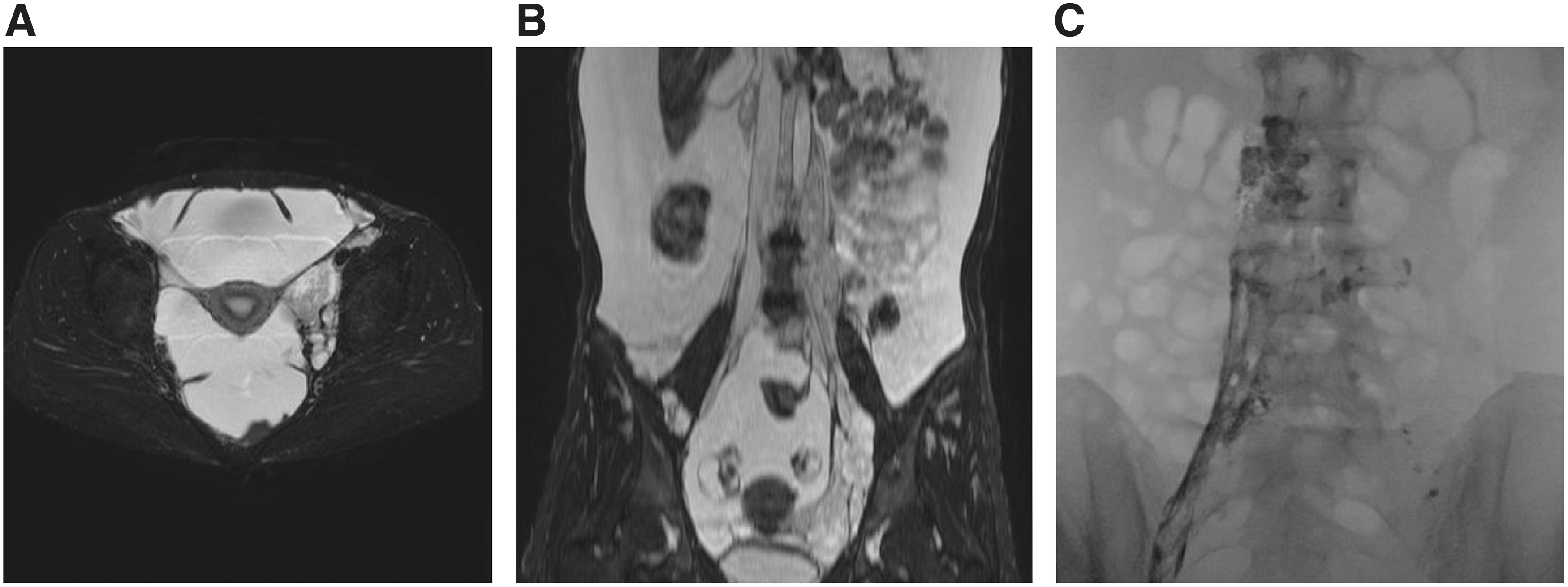
The radiological findings in patients with GLA before sirolimus treatment.
The radiological findings in patients with GLA after sirolimus treatment.
The optimal dose of sirolimus remains controversial. Currently, there are no standardized methods for its optimal dosing. A previous study demonstrated that the highest dose of rapamycin had an effect of reducing the incidence of lymphangiectasia, but it might increase toxicities. 61 A systematic review of sirolimus treatment for vascular anomalies also reported that the expected trough levels of sirolimus for most studies (19/25, 76.0%) were 5–15 ng/mL. 62 Although these diseases often occur in childhood, data for pediatric patients are very limited. However, population modeling study of sirolimus and recommended dosing for infants was reported by the pharmacological group of Cincinnati Children's Hospital Medical Center. 63 –65 Notably, some articles reported the potential risk of severe side effects (including Pneumocystis carinii pneumonia and other infections) in infants treated with sirolimus. 66,67 When pediatric patients are treated with sirolimus, we should therefore simultaneously perform Pneumocystis prophylaxis and monitor the trough levels for safe use. Further clinical trials are important to establish the optimal dosing method of sirolimus, although at present, many prospective clinical trials are ongoing. For example, we are currently undertaking a single-arm, multicenter, prospective study on sirolimus treatment for intractable lymphatic anomalies (SILA study) (UMIN000028905).
In 2018, Venot et al. demonstrated that a PIK3CA inhibitor potentially improved the symptoms of patients with PROS. 68 This was the first evidence suggesting that PIK3CA inhibition is a promising therapeutic strategy in such patients. It is anticipated that the development of such targeted therapies will accelerate in the future.
If pharmacotherapy fails for patients, surgical treatment or radiotherapy should be considered (Fig. 11). The patient with chylothorax undergoes thoracentesis and pleural drainage to improve respiratory distress. If conservative treatment fails, thoracic duct ligation may be effective for pleural effusion. If lymphangiography or DCMRL demonstrates thoracic duct dysfunction, thoracic duct embolization may have a role in improving the patient's status. 69 Lymphaticovenous anastomosis might also be a therapeutic option for intractable lymphatic pleural effusion or ascites. 70
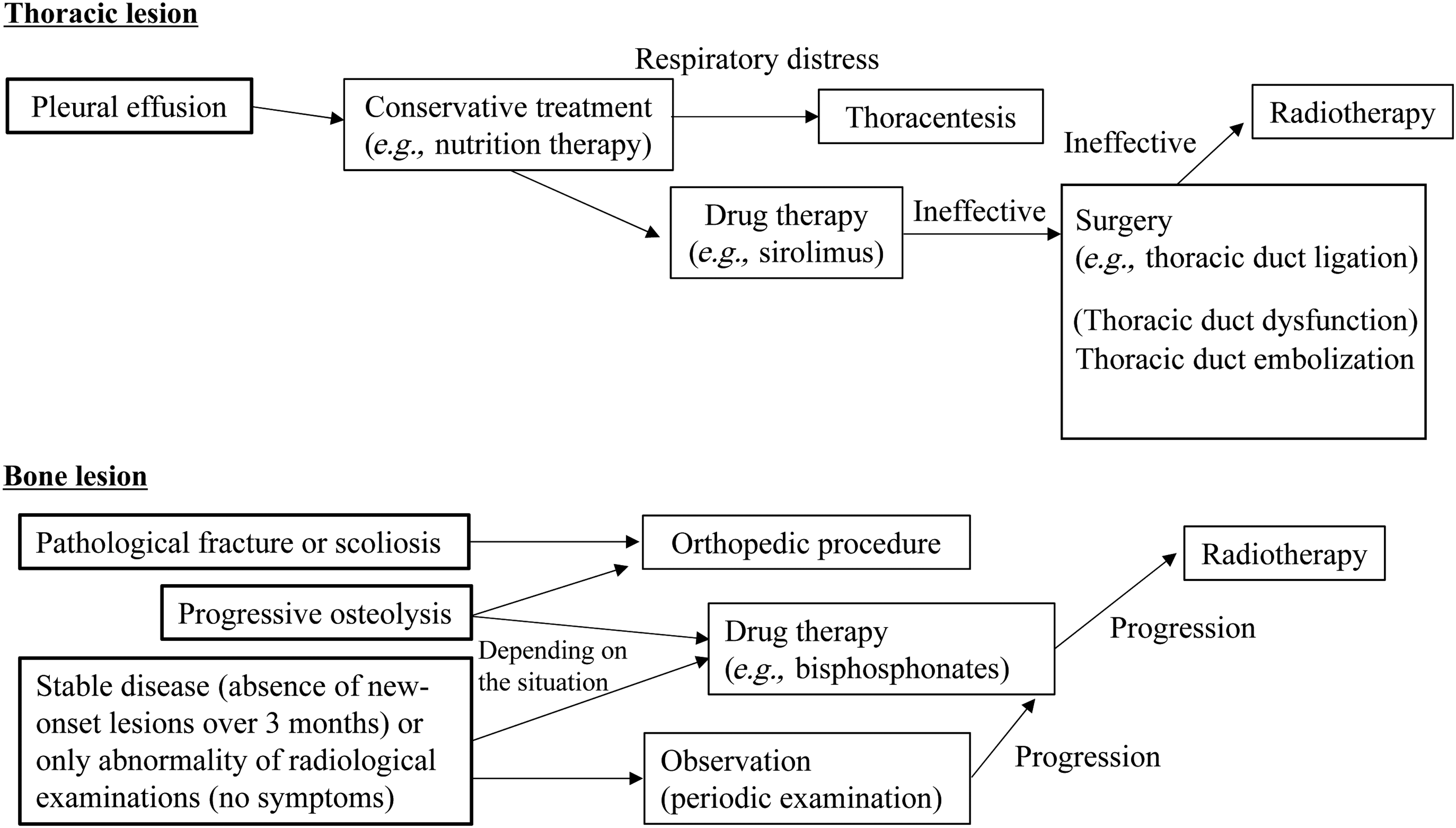
Flow chart of the treatment algorithm for lymphatic anomalies. This flow chart shows the course of treatment for thoracic and bone lesions of lymphatic anomalies. These are only examples and appropriate treatment consistent with individual conditions is necessary.
If surgery is not possible, radiotherapy is an alternative treatment, independently or simultaneously. A previous study showed that radiation therapy had excellent efficacy in stabilizing disease in ∼80% of GSD patients. 71 Dunbar et al. concluded the patients who were treated with intermediate dose of 40–45 Gy with fractionated radiotherapy. 72 However, these doses can be associated with the risk of heart and lung radiation hazards due to radiation exposure to the chest. It is known that doses in the 36–45 Gy range have a reliable effect, but further lower doses (16–20 Gy) also appear to be able to relieve thoracic symptoms (chylothorax and chylopericardium). Radiotherapy for pediatric patients may cause other late adverse effects on bone growth disturbance and secondary malignancy; however, cases that do not improve after other treatments cannot avoid radiotherapy to relieve their symptoms. If the patient has lesions of the chest wall pleura and lung parenchyma, they should be treated with radiotherapy of the thorax to prevent crisis in respiratory symptoms.
Summary
CLAs have extensive and complex lesions, characterized by an abnormal lymphatic system and overlapping clinical symptoms. The patterns of radiological findings of osteolytic lesions can be used to distinguish between GLA/KLA and GSD, along with the presence of infiltrative, progressive bone resorption in GSD patients. In contrast, typical GLA bone lesions are multiple lytic osteolysis. Facilitating the differential diagnosis, KLA patients have a distinctive pathological hallmark, kaposiform hemosiderotic, spindled-shaped lymphatic endothelial cells. Lymphangiography and DCMRL can help to detect abnormalities of the thoracic duct and lymphatic flow in CCLA. Although it remains challenging to diagnose and treat CLAs, the mTOR inhibitor sirolimus could offer a breakthrough for these intractable diseases. In molecular research, data from comprehensive genetic analyses on vascular anomalies allow further insight into their pathogenesis. Pathological animal models also help to deepen our knowledge of the etiology of these diseases. Somatic activating mutations within the genes encoding components of the PIK3/AKT/mTOR and RAS/MAPK pathways have recently been detected in patients with CLAs and numerous vascular anomalies. Identification and characterization of these genetic abnormalities are changing the paradigm by which lesions are diagnosed, potentially leading to novel target therapies and biomarkers.
CLAs are intractable lymphatic disorders, and include GLA, GSD, KLA, and CCLA. Their pathogenesis is unknown and their differential diagnosis remains difficult due to their similar clinical findings.
KLA is a subtype of GLA that is characterized by progressive and severe features (severe thrombocytopenia, consumptive coagulopathy, and bleeding), so an accurate diagnosis is vital.
Osteolytic imaging can distinguish between GLA/KLA and GSD.
Lymphangiography and DCMRL can facilitate the identification of abnormal structure of lymphatics vessels or channels, which may be responsible for leakage of lymph fluid.
Sirolimus might have efficacy in improving the symptoms of patients and could be a novel first-line therapy for CLA patients.
The genes encoding components of the PIK3CA and RAS pathways are associated with the pathology of CLAs, understanding of which can deepen our knowledge of diseases and lead to novel target treatments.
Footnotes
Acknowledgments and Funding Sources
This study was supported by a grant from the Practical Research Project for Rare/Intractable Diseases from Japan's Agency for Medical Research and Development, AMED (18lk0201055h0003); and a Health and Labour Science Research Grant for Research on Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan received by M.O. We thank the cooperating institutions in Japan and Ms. Asuka Ogawa, Dr. Akifumi Nozawa, Dr. Shiho Yasue, Dr. Saori Endo, Dr. Yukichi Tanahashi, Dr. Tomohiro Kanayama, Dr. Kazuhiro Kobayashi, Dr. Natsuko Suzui, and Dr. Tatsuhiko Miyazaki, Gifu University, for helpful comments and data review. We also thank the staff at the Department of Pediatrics at Gifu University for their contribution. We thank Simon Teteris, PhD, from the Edanz Group, for editing the English text of a draft of this article.
Author Disclosure and Ghostwriting
The authors declare that they have no competing financial interests to disclose. The content of this article was expressly written by the authors listed and no ghostwriters were used.
About the Authors