
Abstract
Objective:
Fetuses early in gestation heal skin wounds without forming scars. The biological mechanisms behind this process are largely unknown. Fibroblasts, however, are cells known to be intimately involved in wound healing and scar formation. We examined fibroblasts in different stages of development to characterize differences in gene expression that may result in the switch from regenerative wound repair to repair with scarring.
Approach:
Fibroblasts were isolated and cultured from the back skin of BALB/c wild-type mouse fetuses at embryonic day (E)14 and E18 (n = 10). The fibroblast total RNA was extracted, and microarray analysis was conducted using chips containing 42,000 genes. Significance analysis of microarrays was performed to identify genes with greater than twofold expression difference and a false discovery rate of less than two. Identified genes subsequently underwent enrichment analysis to detect differentially expressed pathways.
Results:
Two hundred seventy-five genes were differentially expressed between E14 and E18 in fetal fibroblasts. Thirty genes were significantly downregulated and 245 genes were significantly upregulated at E18 compared with E14. Ingenuity pathway analysis identified the top 20 signaling pathways differentially activated in fetal fibroblasts between the E18 and E14 time points.
Innovation:
To our knowledge, this work represents the first instance where differentially expressed genes and signaling pathways between fetal fibroblasts at E14 and E18 have been studied.
Conclusion:
The genes and pathways identified here potentially underlie the mechanism behind the transition from fetal wound healing via regeneration to wound healing by repair, and may prove to be key targets for future therapeutics.

Introduction
T
Interestingly, Rowlatt described the ability of the human fetus, early in gestation, to repair cutaneous wounds without forming a scar. 2 Further work has subsequently confirmed these findings in both animal models and in human fetuses. 3 The finding of scarless wound healing in human skin suggests there is potential that adult skin can be stimulated to function as fetal skin, and heal via regeneration rather than fibrosis. The mechanisms of scarless healing, however, require further characterization before such knowledge can be applied for therapeutic benefit.
There are numerous differences documented between fetal and adult wound healing phenotypes. Scarless wound healing is age dependent; there is a distinct switch from regenerative to scarring healing around 24 weeks of gestation in human embryos 4 and around gestational age 18 days (embryonic day [E]18) in mice. 5 Scar-free healing is also dependent on the size of the wound, with larger wounds healing via scarring at earlier gestational ages. 4 In vitro, fetal skin fibroblasts are able to simultaneously proliferate and synthesize collagen, whereas adult skin fibroblasts proliferate before they synthesize collagen, suggesting a central role of fibroblasts in the mechanism of scarless wound healing. 6 This study focused on the difference in fibroblast function at different ages of gestation. A microarray transcriptional profiling comparison was conducted on fetal fibroblasts harvested from mouse fetuses at E14 and E18 to detect any deviations in transcriptomes between scarless and scarring repair. The aim was to identify novel pathways involving fibroblasts that promote regenerative repair and scarless healing.
Clinical Problem Addressed
Scarring is the expected outcome of the human adult wound healing process and is a significant medical issue that can substantially reduce patients' quality of life. Numerous physiological and psychological consequences result from scarring, either the result of trauma or surgery. 7 Facial scars are often cosmetically displeasing and result in substantial psychosocial distress. Keloid scars can cause immense pain and severe itching, while hypertrophic scars can lead to contracture, erosion of skeletal structure, and potentially lifelong disability. 8 Scars and their associated consequences surmount to enormous economic cost estimated in the tens of billions of dollars. 9
Materials and Methods
Animals
BALB/c wild-type mice at 6 weeks of age were purchased from Charles River Laboratories (Wilmington, MA). After acclimation for at least 1 week, male and female mice were bred overnight. Every day, the female mice were checked and the day of vaginal plug was determined to be E0.5 for gestational timing. All animal procedures were performed in accordance with National Institutes of Health (NIH) guidelines according to university-approved protocols. Mice were closely monitored by the Stanford Administrative Panel on Laboratory Animal Care (APLAC).
Primary cell culture
Gestational age E14 and E18 pregnant mice were sacrificed with CO2 and cervical dislocation. Fetal mice were surgically removed using sterile technique. Under a dissecting microscope, E14 (n = 10) and E18 (n = 10) fetal mouse dorsal skin was collected and pooled for fibroblast isolation and primary cell culture under sterile conditions.
Fibroblast primary cell culture was conducted by mincing tissue and treating it with 0.25% trypsin/ethylenediaminetetraacetic acid (EDTA) in 37°C with mild agitation for 10 min. Mouse embryonic fibroblast culture medium consisting of Dulbecco's modified Eagle's medium, GlutaMAX supplement (Thermo Fisher Scientific, Waltham, MA), 10% fetal bovine serum (Omega Scientific, Tarzana, CA), 0.1 mM 2-mercaptoethanol (Sigma, St. Louis, MO), and 1% penicillin/streptomycin (Thermo Fisher Scientific) was used for cell culture. Cells were cultured at 37°C in a humidified incubator with 5% CO2. All experiments used passage one fibroblasts from male and female fetuses.
RNA extraction and amplification
RNA was extracted using the TRIzol protocol (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Subsequently, the MessageAmp antisense RNA (aRNA) kit (Ambion, Austin, TX) was used to amplify 1 μg of extracted RNA from each group. To compare the different arrays between gestational time points, universal mouse RNA was amplified in individual reaction mixtures of 1 μg aliquots simultaneously, with amplifications in the experimental sample and utilized as an internal amplification control.
Preparation of fluorescent complementary DNA probes
Two micrograms of random hexamer and 4 μg of RNA were heated at 65°C for 10 min. Samples were then reverse transcribed with 2 mM of each deoxyribose nucleotide triphosphate, 1 × first-strand buffer, 0.5 μL RNAse inhibitor, 200 U superscript II, 10 mM dithiothreitol, and either 3 μL Cy3-deoxyribose uridine triphosphate (dUTP) (for experimental samples) or Cy5-dUTP (for universal mouse control samples) at 42°C for 1 h in a 30 μL total reaction volume. To increase the reaction, 200 U superscript II was added to the mixture and samples were incubated at 42°C for 1 h. Fluorescent Cy3- or Cy5-labeled probes were washed with Tris-EDTA (TE) buffer (10 mM Tris, 1 mM EDTA) through a microcon mini column (Millipore, Billerica, MA), treated with 450 μL TE buffer, and the inverted mini column was spun into a new tube. Microarray chips were immediately hybridized with probes.
Pretreatment of microarray chips
The Stanford Microarray Database Center was used to print microarray chips with 42,000 specific complementary DNAs (cDNAs) printed onto lysine-coated slides. Sequences and accession numbers can be accessed at
Microarray hybridization
Fluorescent-labeled probes were heated at 100°C for 2 min, denatured, and then incubated at 37°C for 20 min. The recovered probe hybridization mixture with a volume of 32 μL, 6.8 μL of 20 × saline sodium citrate (SSC), and 1.2 μL of 10% sodium dodecyl sulfate (SDS) was placed onto microarray slides that were prewarmed. Coverslips were applied and slides were placed into a sealed moisture chamber at 65°C for 16 h to hybridize. Slides were then immediately washed with 1 × SSC in 0.03% SDS, with 0.5% SSC twice, and with 0.06% SSC twice. After the washes, slides were centrifuged at 84 g for 2 min and immediately scanned. An Axon microarray scanner (Molecular Devices, Sunnyvale, CA) was used for scanning slides.
Microarray data analysis
GenePix Pro 4.0 software (Molecular Devices) was used to analyze scanned slides. Densitometry data for gene identification and analysis were uploaded into the Stanford Microarray Database. The log (base 2) of red/green normalized ratio (mean) was found and filtered based on a regression correlation of 0.6. Each gene was centered to the median and was only included in the final analysis if genes passed >80% good data filter criterion. Pearson correlation was used to cluster genes. Subsequently, genes between E14 and E18 with significant differences were selected using significance analysis of microarrays (SAM). By utilizing a set of gene-specific t tests, SAM is able to identify gene expression changes that are statistically significant. The analysis assigns a score to each individual gene dependent on the expression change based on the standard deviation of the gene repeated measurements. Permutations of repeated measurements are used to determine the false discovery rate (FDR) for genes equivalent to chance. Only genes that had both an FDR less than two and at least twofold expression difference were selected.
Functional analysis of differentially expressed genes
As previously described by Jovov et al.,
10
network and pathway analyses of probes were performed using Ingenuity Pathway Analysis (IPA;
Results
Differential gene expression between scarless E14 and scarring E18 fibroblasts
SAM identified a total of 275 genes that were differentially expressed, with at least a twofold difference, in fetal fibroblasts between E14 and E18. Of these genes, 30 were significantly downregulated (Table 1) and 245 genes were significantly upregulated at the E18 compared with the E14 time point (Table 2).
Genes downregulated in E14 fibroblasts
Genes upregulated in E14 fibroblasts
Functional pathway analysis
Of the 245 genes upregulated in E18 compared with E14 fetal fibroblasts identified using microarray analysis (Fig. 1A), IPA identified 20 functional pathways represented by these genes (Fig. 1B). The top five pathways identified were associated with colorectal cancer (CRC) metastasis signaling, hepatic fibrosis/hepatic stellate cell activation, pancreatic adenocarcinoma signaling, platelet-derived growth factor (PDGF) signaling, and endometrial cancer signaling. From the 30 genes downregulated in E18 compared with E14 fibroblasts, IPA identified 20 functional pathways represented by these genes (Fig. 1C). The five most significant functional pathways identified were putrescine biosynthesis III, s-adenosyl-l-methionine biosynthesis, d-myo-inositol (1,4,5,6)-tetrakisphosphate biosynthesis, d-myo-inositol (3,4,5,6)-tetrakisphosphate biosynthesis, and dolichyl-diphosphooligosaccharide biosynthesis.
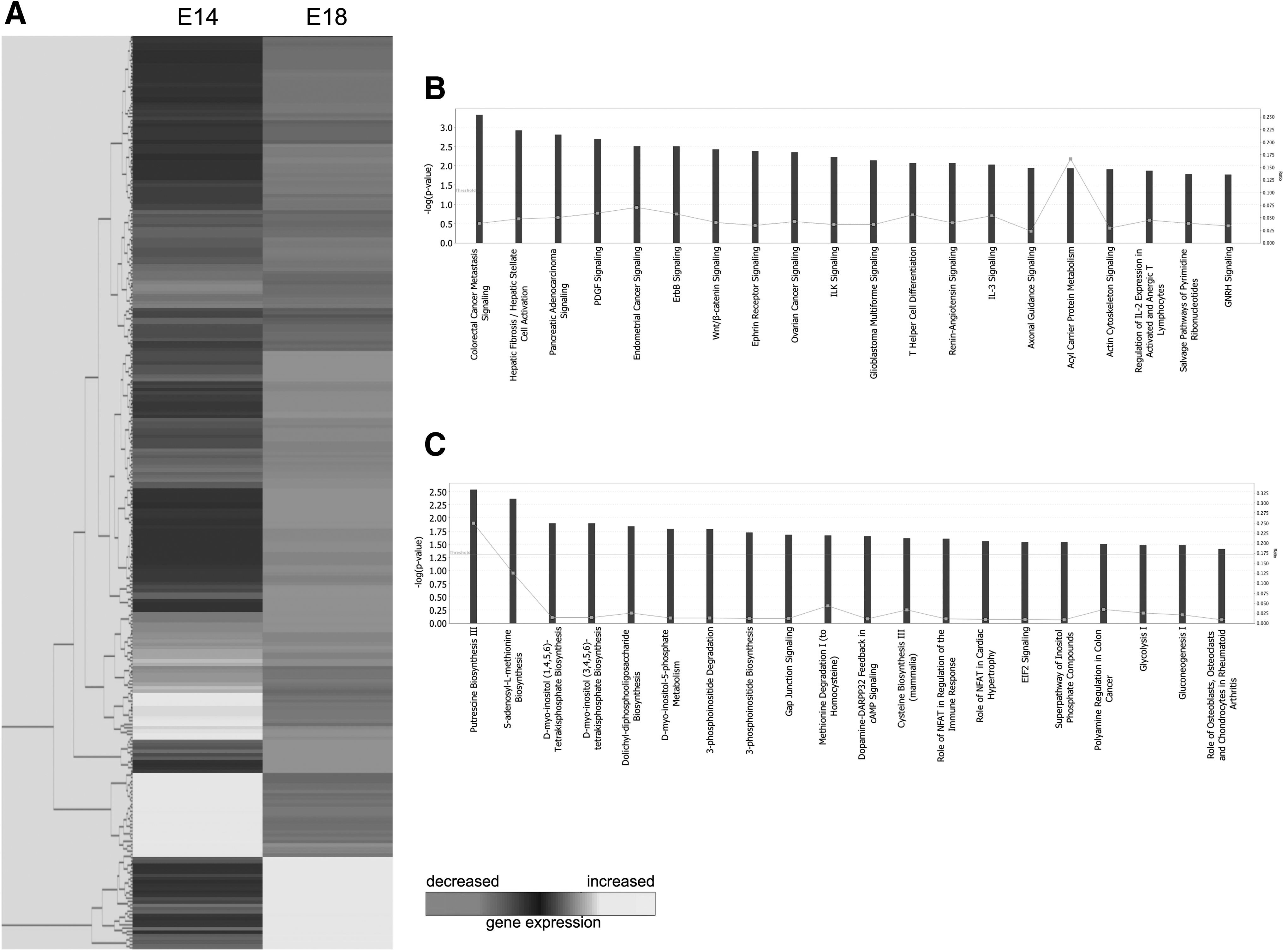
Microarray analysis of E14 and E18 fibroblasts.
Discussion
Previous work conducted by our laboratory analyzed the differences in the transcriptomes of fetal keratinocytes and fibroblasts between E16 and E18. At the time of this research, however, it was difficult to efficiently culture E14 fetal fibroblasts. 11 Using a larger number of E14 fetal mice and a smaller culture plating surface in this study, we were able to successfully culture E14 fetal fibroblasts. The microarray analysis and signal pathway analysis conducted at the E14 time point in this study revealed a number of significant gene expression changes that occur during the transition period from scarless healing to healing with scarring. A recent investigation by our laboratory compared gene expression in fetal (E17) and adult mouse wounds and identified 178 genes upregulated and 13 genes downregulated in fetal compared with adult wounds. A selection of down- and upregulated pathways was identified (unpublished observations). Together with the findings presented here, this research contributes to unraveling the mechanism of fetal regenerative cutaneous wound repair. In the following sections, we characterize in greater detail some of the notable pathways differentially activated in E14 fibroblasts versus E18 fibroblasts.
CRC metastasis signaling
SAM revealed a significant increase in CRC metastasis signaling in E18 compared with E14 fetal fibroblasts. CRC tumorigenesis is well characterized and results from the accumulation of genetic mutations. Inflammation is a known environmental factor strongly associated with genetic mutations in cells of the colonic mucosa, which predisposes it to developing CRC. The association between CRC tumorigenesis and inflammation is demonstrated by the increased risk of CRC in patients with extensive and long-term colitis. 12 A number of proinflammatory factors are activated by the network of CRC signaling, 13 including TNFα, interleukin (IL)-8, IL-6, and VEGF, which are all found to be elevated in the serum of patients with CRC. 14 Strong evidence identifies transforming growth factor-β (TGFβ) as one of the key factors promoting inflammation during tumorigenesis. 15 Interestingly, inflammation and TGFβ have also been demonstrated to play an immediate role in the fibrotic response to cutaneous injury, and TGFβ expression is significantly elevated in the fibroblasts present in keloid and hypertrophic scars. 16 It is highly likely, therefore, that the observed upregulation of CRC metastasis signaling in fetal fibroblasts at E18 compared with E14 is associated with increased levels of proinflammatory factors, including TGFβ, which ultimately contribute to scar formation. Consequently, targeted inhibition of one or more proinflammatory factors upregulated in scar-forming fetal fibroblasts could significantly improve the wound repair process in adult cutaneous wounds.
Hepatic fibrosis/hepatic stellate cell activation
A significant upregulation of hepatic fibrosis/hepatic stellate cell activation pathway in fetal fibroblasts at E18 compared with E14 was found. This pathway involves activation of the hepatic stellate cell following liver injury, the principal effector of hepatic fibrogenesis, which becomes highly proliferative and synthesizes a fibrotic matrix that is rich in type I collagen. 17 Hepatic fibrosis can arise secondary to a number of factors causing liver injury, including viral hepatitis or alcohol abuse. Regardless of the precise etiology, hepatic fibrosis is ultimately characterized by an increase in ECM deposition and formation of a hepatic scar. Immediately following liver injury, there is a marked transition of hepatic stellate cells from a quiescent to a robust, activated state. This response is elicited by neighboring Kupffer, endothelial, and injured hepatocyte cells, which release potent reactive oxygen intermediates (ROI) that exert paracrine stimulation of stellate cells. 18 Excessive accumulation of reactive oxygen species (ROS) in wounds significantly impairs wound healing and causes substantial tissue damage. 19 Our findings here suggest a differential stellate cell activation pathway between the E14 and E18 time points in fetal fibroblasts, and this could be partially caused by increased ROS in the wound bed at E18. Accordingly, methods that eliminate accumulated ROS in wound beds may enhance the regenerative capability of skin.
In addition to activating stellate cells, endothelial cells also activate plasmin. Plasmin stimulates the conversion of latent TGFβ1 to an active fibrogenic form. 20 TGFβ, as mentioned, plays a major role in the fibrotic response to injury and the formation of scars. Therefore, targeted inhibition of the conversion of TGFβ1 into its more active fibrogenic form will likely enhance the regenerative properties of skin once beyond the E14 time point.
PDGF signaling
Our microarray analysis demonstrated significantly increased expression of PDGF in E18 compared with E14 fetal fibroblasts. Higher levels of PDGF have been documented both in adult wounds compared with embryonic wounds, and in uninjured adult skin compared with uninjured embryonic skin. 21,22 No previous work has specifically compared the differential activation of PDGF signaling between early scar-free fetal skin and later scar-forming fetal skin. The functional pathway analysis here suggests that increased PDGF signaling contributes to the loss of the regenerative healing capability between the E14 and E18 time points. This finding is further supported by the role of PDGF in mediating proliferation and differentiation of fibroblasts. PDGF expression also leads to subsequent upregulation of TGFβ1 receptors, which have a major role in the fibrotic response to tissue injury.
Putrescine biosynthesis III
Microarray analyses showed greater downregulation of putrescine biosynthesis in E18 compared with E14 fetal fibroblasts. Putrescine has been identified to play a significant role in cell proliferation in response to injury, and is required for the completion of DNA synthesis. 23 Putrescine is also suggested to be an integral precursor for the development of complex polyamines, which are major regulatory factors of mammalian tissues and influence both growth and signal transduction. 24 The formation of new tissue, through cellular proliferation and migration of cells to the wound site, is a key part of wound healing. Downregulation of the putrescine biosynthesis III pathway at E18 may impair signaling, impact the regulation of cellular proliferation, and ultimately increase the amount of new tissue formed, leading to the formation of a scar. Further exploration of the putrescine biosynthesis III pathway could reveal a target for drug therapy that could potentially rescue regenerative capability at scar-forming time points.
D-myo-inositol (3,4,5,6)-tetrakisphosphate biosynthesis
Microarray analysis revealed that d-myo-inositol (3,4,5,6)-tetrakisphosphate biosynthesis was downregulated in E18 compared with E14 fetal fibroblasts. D-myo-inositol (3,4,5,6)-tetrakisphosphate is part of the inositol phosphate family, which are intracellular signaling molecules regulating the crucial functions of cell growth, apoptosis, and cell differentiation. Previous work has tentatively identified the target of d-myo-inositol (3,4,5,6)-tetrakisphosphate as a plasma membrane Ca2+-activated chloride channel. 25 Interestingly, blockage of chloride channels severely reduces the rate of wound healing. 26 Downregulation of chloride channel function at E18 may reduce healing capability in fetal wounds and prolong the formation of new tissue and scars. D-myo-inositol (3,4,5,6)-tetrakisphosphate is also involved in mediating the ability of remodeling complexes to induce transcription in phosphate-responsive genes. 27 This finding suggests that reduction in d-myo-inositol (3,4,5,6)-tetrakisphosphate activity may contribute to epigenetic silencing and consequently the loss of regenerative healing capability observed at E18. Further work with d-myo-inositol (3,4,5,6)-tetrakisphosphate biosynthesis could reveal a novel target to induce regenerative healing mechanisms.
Innovation
Previous work has identified the key time point during fetal development when the ability to heal without scar is lost. However, the mechanism driving the transition from scarless, regenerative fetal wound healing to scar-forming adult wound repair is still largely unknown. Our work significantly narrows the search to a set of key genes and pathways that drive the scar-free healing mechanism. Further detailed examination of these genes and signaling pathways will give us a better understanding of the mechanism behind scarless wound healing and potentially identify targets for future therapies.
Footnotes
Acknowledgments and Funding Sources
This work was supported, in part, by an NIH grant R01 GM087609 (to H.P.L.), a gift from Ingrid Lai and Bill Shu in honor of Anthony Shu (to H.P.L.), and the Hagey Laboratory for Pediatric Regenerative Medicine and Children's Surgical Research Program (to M.T.L. and H.P.L.). Additional funding was provided by the American Society of Maxillofacial Surgeons (ASMS)/Maxillofacial Surgeons Foundation (MSF) Research Grant Award (to M.S.H., M.T.L., and H.P.L.), the Sarnoff Cardiovascular Research Foundation (to W.X.H.), the California Institute for Regenerative Medicine (CIRM) Clinical Fellow training grant TG2-01159 (to M.S.H.), and the Stanford University School of Medicine Transplant and Tissue Engineering Fellowship Award (to M.S.H.).
Author Disclosure and Ghostwriting
No competing financial interests exist. The content of this article was expressly written by the authors listed. No ghostwriters were used to write this article.
About the Authors