
Abstract
Nucleic acid (NA) sensing receptors were first described in the context of host defense. We now know that some endosomal NA sensors play a critical role in the development of systemic autoimmune diseases such as systemic lupus erythematosus, whereas cytosolic Cyclic GMP-AMP Synthase/Stimulator of IFN Genes (cGAS/STING) DNA-detecting pathway has been associated with monogenic autoinflammatory interferonopathies such as Aicardi–Goutieres and Education; collaboration; communication STING-associated vasculopathy with onset in infancy (SAVI). DNaseII hypomorphic patients and DNase−/− IFNaR−/− (double knockout [DKO]) mice also develop an autoinflammatory syndrome associated with an interferon signature. We now add to the description of an unusual clinical manifestation of DKO mice that involves the accrual of trabecular bone in long bone marrow and the formation of ectopic bone within the spleen. This aberrant bone formation is lost not only in STING-deficient but also in Unc93b1-deficient mice and, therefore, depends on the interplay of cells expressing cytosolic and endosomal NA sensing receptors.
Introduction
STING has been implicated in numerous autoinflammatory diseases as shown by the impact of STING deficiency on these conditions. We now show that Unc93b1-deficiency can have an equally dramatic effect on certain clinical manifestations of DNaseII-deficient mice. These data point to a potentially critical interplay between cytosolic and endosomal nucleic acid sensing receptors in autoinflammatory diseases.
The immune system has devised remarkable strategies to maintain host defense and at the same time prevent potentially pathogenic self-reactivity. In the case of T cell viral immunity, this discrimination is maintained by recognition of self major histocompatibility complex in combination with a virally derived peptide, as elegantly shown by Peter Doherty and Rolf Zinkernagel in their Nobel prize-winning studies (30,31). This kind of rigorous hypothesis-based experimentation was the standard that Peter passed on to his trainees and that I have tried to emulate throughout my career. Another lesson learned from Peter was to follow the data and that has led me from T cell immunology to innate immunity!
In the world of innate immunity, a critical aspect of host defense is provided by the pattern recognition receptors that detect microbial products and trigger antigen presenting cells to present their peptide epitopes in an immunogenic context (18,26). Intriguingly, studies for the past two decades have further shown that many of these receptors, first identified by their ability to detect pathogen-associated molecular patterns, also detect danger-associated molecular patterns that result from sterile inflammation (10,22). These receptors have now emerged as key players in the initiation and pathogenesis of autoimmune and autoinflammatory diseases, and include endosomal and cytosolic nucleic acid (NA) sensing receptors that have been widely studied by many laboratories, including my own.
NA sensing receptors were first described in the context of host defense where they detect the aberrant accumulation of cytosolic or endosomal DNA or RNA resulting from microbial infection (16,19,25,27). Distinct families of germline-encoded pattern recognition receptors sense both microbial as well as endogenous NAs and other danger signals released from stressed, damaged, or dying cells (1,17,22). As originally described, endosomal Toll-like receptors (TLRs) play a critical role in murine models of systemic lupus erythematosus (SLE) and the associated autoantibody production (14,15). In addition, failure to appropriately control the level of autologous DNA or RNA in the cytosol can lead to the activation of cytosolic NA sensors and a number of recently described monogenic autoinflammatory diseases (23).
Several cytosolic DNA receptors have been identified. These include AIM2, a DNA sensor that forms an inflammasome capable of processing IL-1b (8), and cGAS in the STING pathway (29). STING plays a critical role in the induction of type I interferon (IFN) and also activates downstream pathways leading to the production of nuclear factor kappa B and the production of proinflammatory cytokines. Importantly, the STING pathway has been implicated as a key sensing mechanism of cytoplasmic DNA accrual in mice and patients that fail to express Trex1/DNaseIII or DNAseII and, therefore, suffer a range autoinflammatory conditions (2,6,7,24). Trex1-deficient patients and mice exhibit nonoverlapping clinical manifestations; the central nervous system is frequently targeted in patients, whereas the heart is targeted in B6 mice. In contrast, there are remarkable similarities between patients with hypomorphic mutations in DNaseII and DNaseII-deficient mice (3,21,24).
DNAseII−/− mice are embryonic lethal, but rescued by deletion of the type I IFN receptor (IFNaR) (28). These DNaseII−/− IFNaR−/− double knockout (DKO) mice survive to adulthood, but develop a late-onset inflammatory arthritis, associated with the production of proinflammatory cytokines, including TNF, IL-1, and IL-6 (12,13,20). Both embryonic lethality and arthritis have been shown to be STING dependent (2). However, based on the nature of the genetic lesion, DNA accrual in phagolysosomes as well as the cytosol, one might anticipate a role for additional NA sensors in disease initiation and/or progression. In fact, we and others have reported that the development of arthritis in DKO mice is also dependent on AIM2 (4,9).
Importantly, DKO mice share additional clinical manifestations of systemic autoimmunity, beyond arthritis, with patients inheriting DNaseII mutations. These include anemia, cytopenia, extramedullary hematopoiesis associated with massive splenomegaly, elevated levels of proinflammatory serum cytokines, the production of antinuclear autoantibodies, and lymphocyte activation, all features that are absent from DNaseII+/− (heterozygous) IFNaR−/− (Het) controls (3,21). These SLE-like autoimmune manifestations are found in very young DKO mice and are dramatically decreased in DKO mice that inherit a functionally inactive form of the endosomal TLR chaperone protein Unc93b1 (Unc93b1 triple knockout, TKO), but not decreased in STING−/− DNaseII−/− IFNaR−/− TKO mice (21). Together these data identify clinical manifestations that depend on either STING (arthritis) or endosomal TLRs (cytopenia, splenomegaly/extramedullary hematopoiesis, and autoantibody production), but not both.
Older DKO mice also develop an inflammatory infiltrate in the bone marrow (BM), and in contrast to the clinical manifestations described earlier, both Unc93b1 TKO and STING TKO mice are rescued from BM inflammation (3). In addition, AIM2, a DNA sensor that induces inflammasome activation, contributes to both splenomegaly and arthritis (4). Therefore, the DKO model provides a unique experimental system for examining synergistic interactions between endosomal and cytosolic NA sensor pathways in systemic inflammatory disease. We now report one more example of the interplay between these receptors.
Materials and Methods
Microcomputed tomography (micro-CT) and radiography were performed and quantified as described previously (5).
Results and Discussion
Inflammatory arthritis is normally associated with systemic bone loss resulting from increased osteoclast generation, in part due to increased levels of TNF, IL-1b, and RANKL. Therefore, in previous studies, we were surprised to find that DKO mice have an unanticipated dramatic increase in trabecular bone formation in long bones. We further identified ectopic bone formation in the spleens of DKO mice, with the development of bone fragments by 7–10 months of age (5). This augmented trabecular bone formation and formation of bone within the spleen was absent in STING TKO mice, implicating STING in this unusual bone phenotype (5). Both the BM and the spleen are sites of erythropoiesis and the DNaseII-deficient mice cannot degrade nuclei extruded from reticulocytes during the end stage of erythropoiesis (11). Therefore, undegraded nuclei are highly likely to provoke inflammation in both the BM and spleen.
Since we had previously shown that BM inflammation was dependent on both STING and Unc93b1 in DKO mice, it seemed reasonable to ask whether functional Unc93b1 is also required for the accrual of trabecular bone and the formation of ectopic bone within the spleen.
We have now analyzed the long bones and spleens from Unc93B1 TKO mice by micro-CT, and compared the results with comparable data collected previously from Het, DKO, and STING TKO mice (5). Quantification of bone parameters included trabecular bone volume/total volume, total bone surface, trabecular connectivity density, trabecular number, and trabecular spacing. As shown previously for the STING TKO mice (5), Unc93b1 TKO mice were completely protected from accrual of trabecular bone when compared with Het and DKO mice (Fig. 1A, B). In addition, the development of ectopic bone in the spleen was also dramatically reduce in Unc93B1-deficient mice when compared with the DKO mice (Fig. 1C), as previously demonstrated in STING TKO (5).
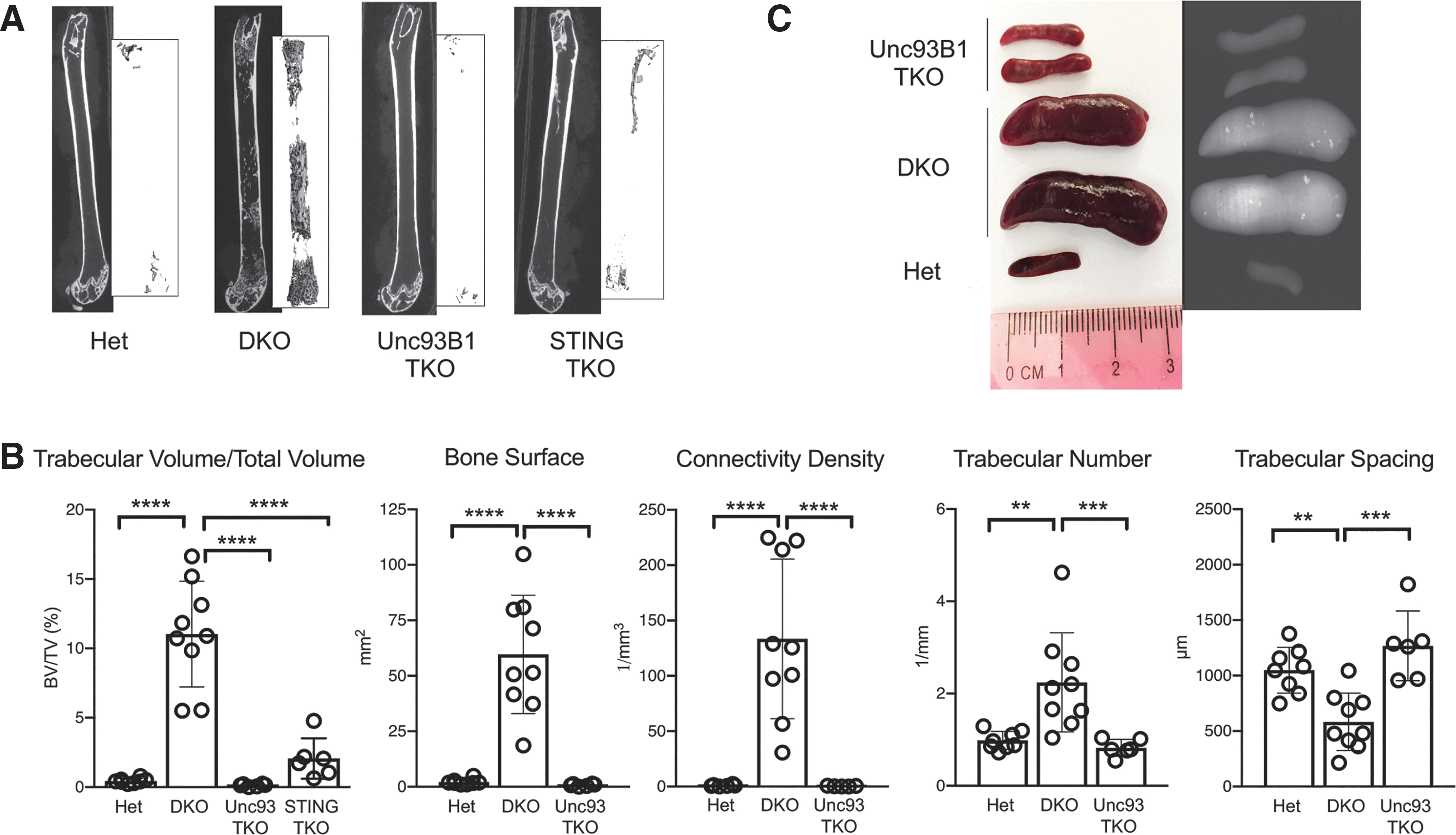
Augmented trabecular bone formation and ectopic bone formation within the spleen is absent in Unc93b1 TKO mice.
The molecular mechanisms responsible for excessive bone accrual remain unresolved and will require further investigation, but it has been shown to reflect increased osteoblast number and function in the long bones. This occurs despite ongoing osteoclast-mediated articular bone erosion and systemically elevated titers of proinflammatory cytokines that under normal circumstances should promote bone loss and not bone accrual. Importantly, these aberrant bone disorders occur late in life (5). By contrast, the Unc93B1-driven clinical manifestations of autoinflammation develop at a much earlier age, leading us to speculate that cell death, resulting from an inability to degrade excessive levels of lysosomal/cytoplasmic DNA by the absence of DNaseII, leads to the activation of Unc93B1-dependent receptors.
This creates an inflammatory environment that then enables STING to subsequently contribute to the final stages of bone formation in the long bones and spleen. Moreover, these data clearly demonstrate that autoinflammation, even in monogenic diseases, can result from the activation of numerous NA sensors. Both patients and DKO mice, but not Unc93b1 TKO mice also develop liver fibrosis (21,24), but the role of STING in liver fibrosis has not yet been explored. A better understanding of the precise pathways activated in DNaseII-deficient cells—macrophages, granulocytes, dendritic cells, osteoblasts, fibroblast, and endothelial cells—could, therefore, reveal important new therapeutic targets for intervention in inflammatory diseases.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Supported by the National Institutes of Health grants T32-AI-095213 to R.B., AR-050256 to A.M.-R., and AR-067394 to E.M.G. Musculoskeletal Center of Excellence Grants Program, UMass Memorial Medical Center grant to E.M.G.