
Abstract
Very little information is available on the role of innate immune mechanisms in Japanese encephalitis virus (JEV) infection. This study was designed to investigate the role of all Pattern Recognition Receptors (PRRs) in JEV infection in a mouse neuronal cell line in comparison to events that occur in vivo, using JEV infected suckling and adult mice. Analysis of mRNA expression was carried out using RT-PCR for detection of PRR genes and their downstream pathway genes, while a PCR array technique was used to examine the complete transcription analysis. Amongst the various innate immune receptors, TLR3 gene exhibited differential expression in JEV-infected Neuro2a, in suckling mice and adult mouse brain cells but not in uninfected control cells. The downstream events of TLR3 were confirmed by increased mRNA expression of IRF3 and interferon-β in JEV-infected Neuro2a cells and suckling mice brain tissue. To confirm the functional significance of this observation, TLR3 gene silencing experiments were carried using specific siRNA in Neuro2a cells. The results revealed a significant enhancement of JEV replication in TLR3 gene silenced JEV-infected Neuro2a cells, thereby suggesting that TLR3 serves a protective role against JEV. The expression levels of other PRRs varied. JEV-infected adult mice showed significant upregulation of TLR2 and MDA5 as compared to JEV-infected suckling mice and Neuro2a cells. In addition, upregulation of Myd88 and IRF7 was also noted in adult mice. These observations, coupled with the fact that adult mice infected with JEV exhibited longer survival rates, suggests that the host antiviral TLR2 response seen in adult mice was eventually countered by the virus by using MDA5 receptor. Our findings suggest that different PRRs appear to be involved in JEV infection in Neuro2a cells and brains of suckling and adult mice.
Introduction
Japanese encephalitis virus (JEV) is a member of the family Flaviviridae and causes an acute encephalitic illness. It is a devastating disease of childhood, endemic to Asia and the Western Pacific. Approximately 67,900 cases are estimated to occur every year in 24 Japanese encephalitis (JE) endemic countries and about 80% of these occur in countries with well established or developing JE vaccination programs (3). The mortality in JE can be as high as 30%, and 50% of survivors suffer severe neurological sequelae, including motor paralysis and cognitive impairment (11). The ratio of symptomatic to asymptomatic infections in JEV infection is estimated to be 1:300–1000. The precise role of innate immune mechanisms in affording protection to the large number of individuals with inapparent infection is not well understood.
The innate immune system plays an important role in response to several viral infections (12). Innate immunity is usually effected through pattern recognition receptors (PRRs), B-cells, and natural antibodies present in the body. The PRRs include toll-like receptors (TLRs) and helicases such as Laboratory of Genetics and Physiology2 (LGP2), retinoic acid inducible gene-I (RIG-I), and melanoma differentiation associated gene 5 (MDA5) (16). TLRs play an important role in innate immunity by sensing a broad range of invading pathogens. Stimulation of TLRs by pathogen-associated molecular patterns (PAMPs), which are highly conserved structural motifs expressed by microbial pathogens, initiates signaling cascades that involve a number of proteins (2,4,10).
Although very little information is available on the role of TLRs in JEV infection, there are reports on the role of TLR3 in West Nile virus (WNV) infection in mice. Wang et al. (21) noted that TLR3 contributed to WNV lethality in mice, while Daffis et al. (7) reported that in TLR3 knockout mice enhanced viral replication was observed in brain, suggesting that TLR3 serves a protective role against WNV (7). Subsequent studies have, however, supported a positive role for TLR3 in protective immunity against WNV (1,22). Delineating the differential roles of MDA5 and RIG-I in recognition of double-stranded RNA viruses, Kato et al. (14) observed that RIG-I is essential for eliciting protection against JEV through a robust interferon production, whereas MDA5 played a similar role in picornavirus infection. These observations, as well as recent studies by Nazmi et al. (17,18) suggest that RIG-I could be a key pathway in controlling JEV infection. Studying the transcriptome profile in the brains of mice following JEV infection, Gupta and Rao (13) noted that there is an increased expression of mRNAs of TLR2, TLR3, and TLR7, suggesting that they have an important role in mediating inflammation and immune response to the virus. None of these studies however have comprehensively addressed the roles of all PRRs in JEV infection, as each has limited their observation to one set of innate immune receptors. Therefore, in this study we investigated the role of all PRRs in JEV infection in vitro, using a mouse neuroblastoma Neuro2a cell line as well as in vivo, by studying JEV infected suckling and adult mice infected through the intracerebral route. Further, we analyzed downstream transcription pathways of all PRRs after JEV infection.
Materials and Methods
Virus
An Indian prototype isolate of JEV, strain P20778, obtained from the National Institute of Virology, Pune, India, was used in the study. Monolayers of Aedes albopictus C6/36 mosquito cell line were grown in T-25 cm2 flasks (Nunc, Denmark). Cells were infected with 0.1 multiplicity of infection (MOI) of JEV and harvested at 72 and 120 h post infection (PI). The titer of the virus (1.5×106) was determined using PS cells, and the same virus stock was used for all the experiments.
Cells
Neuro2a (mouse neuroblastoma cell line) cells were grown in monolayers in T-25 cm2 flask in Dulbecco's modified Eagle's medium (DMEM, Sigma, USA) with 5% FBS (Sigma) and penicillin/streptomycin. Neuro2a cells were seeded at a density of 2×104 cells per well in a 24-well plate (Nunc, Denmark). The cells were infected with 0.1 MOI of JEV and harvested at different time points (18 and 24 h) post infection (PI).
Mice
C56BL6/J strain of suckling mice (4–7 days old) and adult mice (4–6 weeks old) used in the study were obtained from Central Animal Research Facility (CARF), National Institute of Mental Health and Neurosciences (NIMHANS). Institutional Animal Ethics Committee clearance was obtained from the NIMHANS Animal Ethics Committee (IAEC project no. AES/34/223/N.V/April2009).
Inoculation of mice
Suckling mice (n=8) and adult mice (n=8) were inoculated by the intracerebral route by injecting 25 μL of JEV (100 pfu/mouse), while control mice (n=8) were mock infected with 25 μL of DMEM. The mice were observed twice a day for onset of any symptoms. Brains from all the JEV-infected as well as mock-infected mice were harvested, subjected to homogenization, and a 10% suspension was prepared using sterile PBS.
RNA extraction and cDNA synthesis
RNA was extracted from Neuro2a cells infected with JEV at 18 and 24 h PI, uninfected Neuro2a cells as well as from 10% brain tissue suspension of JEV-infected and mock-infected mice using the Tri reagent (Sigma) according to the manufacturer's instructions. The RNA was converted to cDNA using a commercial kit (ABI, USA) and stored at−80°C until use. cDNA concentrations at each time point was matched and adjusted to 1 μg/μL for each reaction in the PCR.
Conventional reverse-transcriptase-PCR
Mouse specific primers for TLRs 1 to 9, RIG-I, and MDA5 genes for conventional PCR were purchased from a commercial source (Invivogen, USA). PCR was carried out using 10 pM of each primer, 10 mM dNTP mix (ABI, USA); 0.25 units of Amplitaq gold (ABI) and 1 μg/μL of cDNA template per reaction were used to obtain products ranging from 300–900 basepairs (bp). The PCR cycling conditions included an initial denaturation step at 94°C for 6 min, followed by 36 cycles of amplification at 94°C for 1 min, 57°C for 1 min, and 72°C for 1 min, followed by final extension at 72°C for 7 min. The PCR products were visualized by electrophoresis on a 2% agarose gel.
Real-time PCR
Real-time PCR for JEV-infected and uninfected Neuro2a cells and JEV-infected and mock-infected suckling and adult mice brain tissues were carried out by using SYBR Green chemistry. Primers for mouse-specific TLRs1-9/RIG-I/MDA5, GAPDH, and JEV-NS5 genes for real-time PCR were designed using sequences from GenBank and Primegen software (Table 1). PCR was carried out using 10 pM of each primer, 12.5 μL of 2x POWER SYBR Green PCR mix (ABI, USA), 8 μL of nuclease free water, and 1 μg of template in a 25 μL reaction. The real-time PCR primers used for detecting mouse PRR cascade pathway genes (IRF3, IRF7, IFNα, IFNβ, IL12, NFκB, and TNFα) were those described earlier (6,15). Identical real-time PCR cycling conditions were used for all assays and consisted of the following steps: initialization at 50°C for 2 min, followed by denaturation at 95°C for 10 min, and 40 amplification cycles at 95°C for 15 sec and 57°C for 30 sec. Finally, melt-curve analysis was done to confirm the specificity of the products. As a positive control for JEV expression in the infected cells/mouse brain tissues, NS5 gene-specific primers were used. GAPDH was used as an internal control (housekeeping gene) and fold changes were calculated for each TLR/RIG-I/MDA5 and cascade pathway genes like IRF3/7, IFNα, IFNβ, and NFκB, which showed differential expression using 2−ΔΔct (23).
PCR array for mouse TLR signaling pathway
RNA extracted from JEV-infected and uninfected Neuro2a cells, as well as JEV-infected and mock-infected mouse brain tissues, was quantitated using Nanodrop (Thermo, USA); 1 μg was converted into cDNA by using a commercial reverse transcription (RT) kit (RT2-first strand kit, Qiagen, USA).
This cDNA was used for mouse-TLR signaling PCR-array (cat. no-PAMM-018A RT2 Profiler PCR array, Qiagen) by using RT2 SYBR Green ROXtm qPCR mastermix (Qiagen) as per manufacturer's instructions. Results were analyzed using SABiosciences PCR Array Data Analysis software (RT2PCR array Profiler) and fold changes in gene expression were calculated.
Indirect immunofluorescence assay
Monolayers of 2×104 of Neuro2a cells were grown in 24-well culture plates and maintained in DMEM (Sigma) with 5% FBS (Sigma). Cells were infected with 0.1 MOI of JEV and fixed with 2% paraformaldehyde (PFA) at 18, 24, and 48 h PI. JEV infection was confirmed by using primary monoclonal antibodies to JEV-E protein and stained with goat anti-mouse secondary antibody tagged with FITC (8). TLR3 expression in Neuro2a cells was studied using anti-TLR3 antibodies (Imgenex, USA) stained with goat anti-mouse FITC conjugate (Genei, USA) and observed under a fluorescence microscope (Nikon).
Confirming the role of TLR3 in JEV-infected Neuro2a cells using siRNA
Neuro2a cells were transfected with TLR3 siRNA specific for mouse (Genesolution siRNA, Qiagen) using a commercially obtained HiPerFect Transfection Reagent (Qiagen) as per manufacturer's instructions. Briefly, 2×104 Neuro2a cells were seeded per well of a 24-well plate in 1 mL/well of DMEM containing serum and antibiotics and incubated at 37°C in a CO2 incubator until 80% confluence of the monolayer was attained. For transfection, 1 μL (10 nM) of each of four siRNA reagents and 6 μL of HiPerFect Transfection reagent were mixed in 90 μL of DMEM to obtain a volume of 100 μL/per transfection, vortexed, and incubated at room temperature for 10–15 min to form transfection complexes. At the end of incubation, 100 μL of transfection mix was added dropwise to the wells of the plate with gentle swirling of the plate to ensure uniform distribution of complexes. The cells were subsequently incubated under their normal growth conditions. At the end of 24 h, the transfection complexes were removed and the cells were either infected with 0.1 MOI of JEV or 1 μg/μLof TLR3 ligand poly I:C(positive control) and incubated at 37°C for 1 h. At the end of incubation, the virus /poly I:C was removed and fresh maintenance medium was added to the cells and incubated for a further period of 18 h. Cells were subsequently harvested and RNA extracted from infected as well as siRNA treated and untreated cells by Tri-reagent method as described above. The RNA was converted to cDNA and real-time PCR was carried out for presence of JEV-NS5 protein gene and TLR3 as described.
Statistical analysis
GraphPad Prism5 software was used to analyze mouse mortality data by log rank (Mantel Cox) test, taking into account both time of death and mortality. P values were generated using Students t-test. A p<0.05 was considered statistically significant. The quantitative real-time PCR data was analyzed by one-way ANOVA, followed by Dunnet's test for comparison between mock and JEV-infected groups. The level of significance was set at p <= 0.05. The real time PCR experiments were repeated twice.
Results
Expression of PRRs in JEV infected Neuro2a cells
TLR1, 2, 4, 5, 6, 7, 8, and 9 genes were detected in both JEV-infected Neuro2a, as well as uninfected Neuro2a cells by conventional PCR (Fig. 1, Panel A). In contrast, TLR3 expression was seen in JEV-infected Neuro2a cells but not in uninfected cells at any time points. RIG-I and MDA5 genes were not expressed in both infected and uninfected Neuro2a cells (data not shown). TLR3 upregulation in JEV-infected Neuro2a cells was subsequently confirmed by relative quantitative PCR. There was a 4.69-fold increase in TLR3 mRNA expression at 18 h PI (Fig. 2).

Agarose gel electrophoresis picture depicting RT-PCR products of TLR1-9 genes from uninfected (control) and JEV infected Neuro2a cells at 18 and 24 h PI (
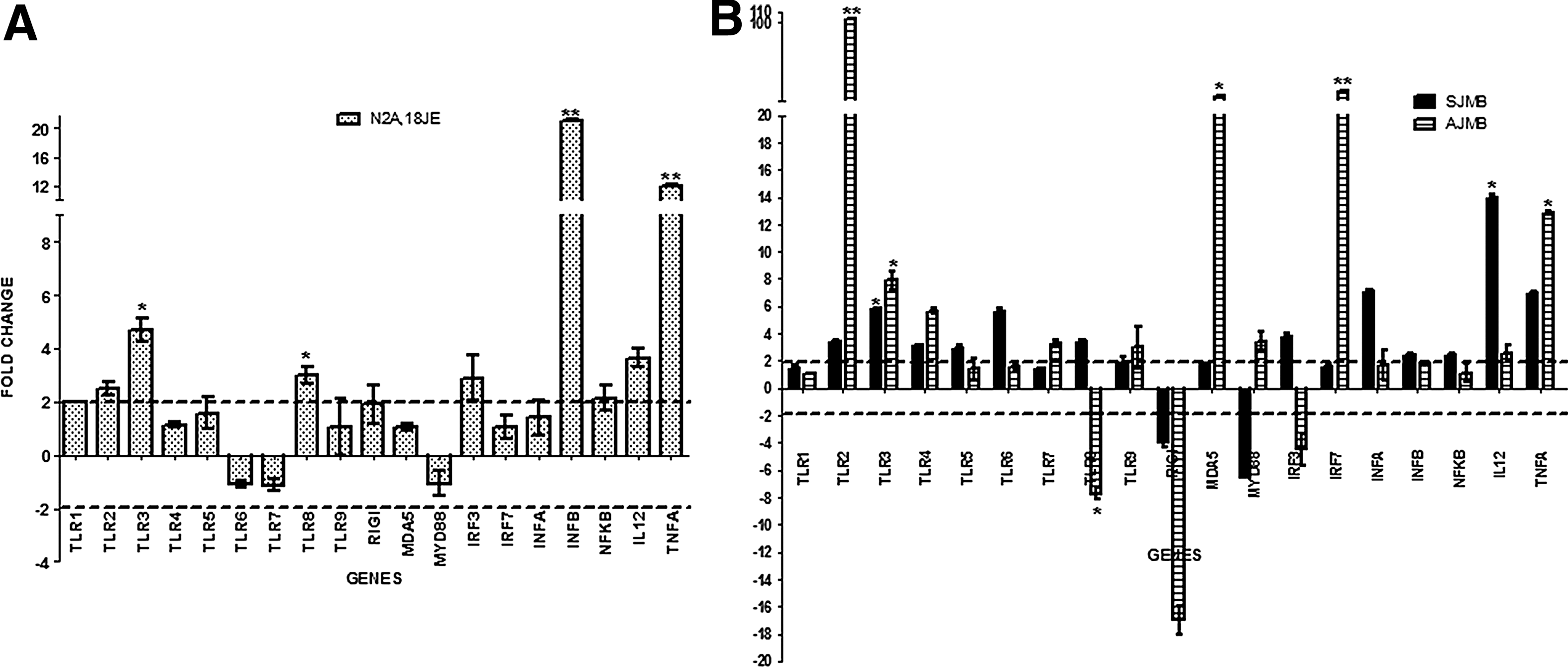
Real Time PCR results depicting fold change in PRRs and cascade pathway genes in JEV-infected Neuro2a cells at 18 h PI (N2A,18JE) are depicted in
PRRs and cascade gene expression by real-time PCR in JEV-infected Neuro2a cells
JEV-infected Neuro2a cells showed upregulation of TLR1, 2, 3, and 8 genes at 18 h PI. Analysis of downstream signaling pathway genes revealed an upregulation of IRF3, NFκB, TNFα, IL12, and IFNβ genes at 18 h PI (Fig. 2).
Detection of TLR3 protein expression in Neuro2a cells
TLR3 protein expression was noted in Neuro2a cells as evidenced by a positive immunofluorescence signal in JEV infected cells as compared to uninfected cells (Fig. 3).
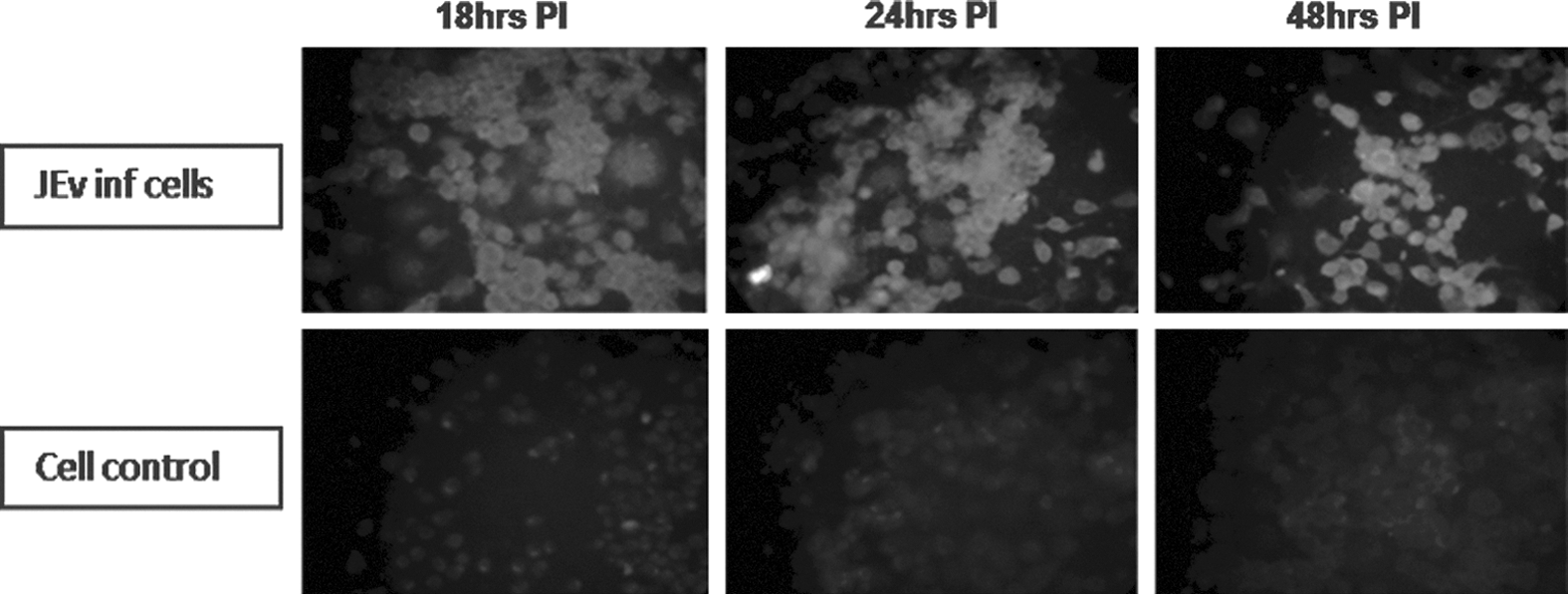
Immunofluorescence assay for TLR3 protein expression after JEV infection in Neuro2a cells at different time points as compared to uninfected cells. Upper panel represents JEV-infected Neuro2a cells at 18, 24, and 48 h PI. Lower panel represents uninfected Neuro2a cells at corresponding time points. Note the bright fluorescence in JEV-infected Neuro2a cells as compared to uninfected cells.
PCR array results of JEV-infected Neuro2a cells
PCR array analysis of JEV-infected Neuro2a cells showed significant changes (Table 2 and Fig. 4) in gene expression up to 18 h post infection (PI). After 24 h PI, no significant change was noted in the expression of these genes. Significant upregulation of TLR3 expression (14.67-fold) was noted and this was further substantiated by upregulation of its downstream cascade genes such as IRF3, IFNβ, and NFκB signaling pathway genes (Table 2, Fig. 4). In addition, a combination of TRAF6 upregulation and Myd88 downregulation was noted, suggesting that there is involvement of TLR3 pathway (Table 2, Fig. 4). Apart from TLR3 activation, there was upregulation of TLRs 2, 5, 6, 7, and 9 gene expressions with downregulation of Myd88. The PCR array results were validated by amplification of selected genes using in-house real time PCR (Supplementary Table S1; supplementary material is available at
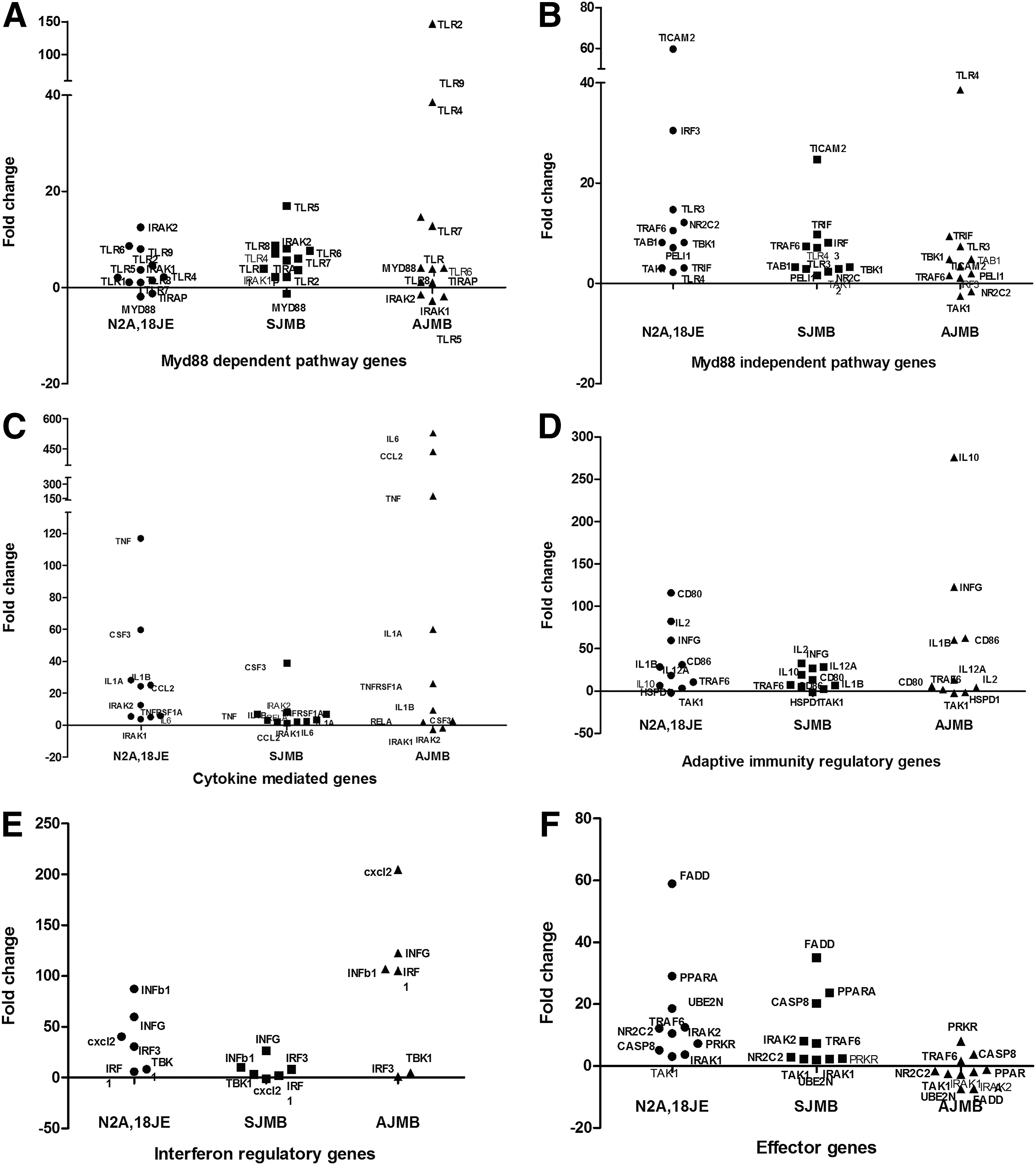
Scatter plots for mRNA expression of different downstream gene pathways activated following JEV infection in Neuro2a cells (N2A,18JE), JEV-infected suckling (SJMB) and adult (AJMB) mice brain tissues by PCR array. In each panel, X axis depicts the cells/brain tissue in which the gene expression was studied, while Y axis depicts the fold change in gene expression in comparison to uninfected control cells/brain tissue.
Genes exhibiting significant upregulation (>2) are depicted in
Mortality pattern of JEV-infected mice
Suckling mice infected intracerebrally with JEV showed initial symptoms of lethargy at 48 h post infection (PI), while in adult mice the initial symptoms were seen at the end of 72 h PI. All suckling mice infected with JEV died after 72 h, while all adult mice succumbed to sickness at 120 h PI. This difference was statistically significant (p=0.0092). The mock-infected mice did not show any symptoms in any of the group at any time point (Fig. 5).
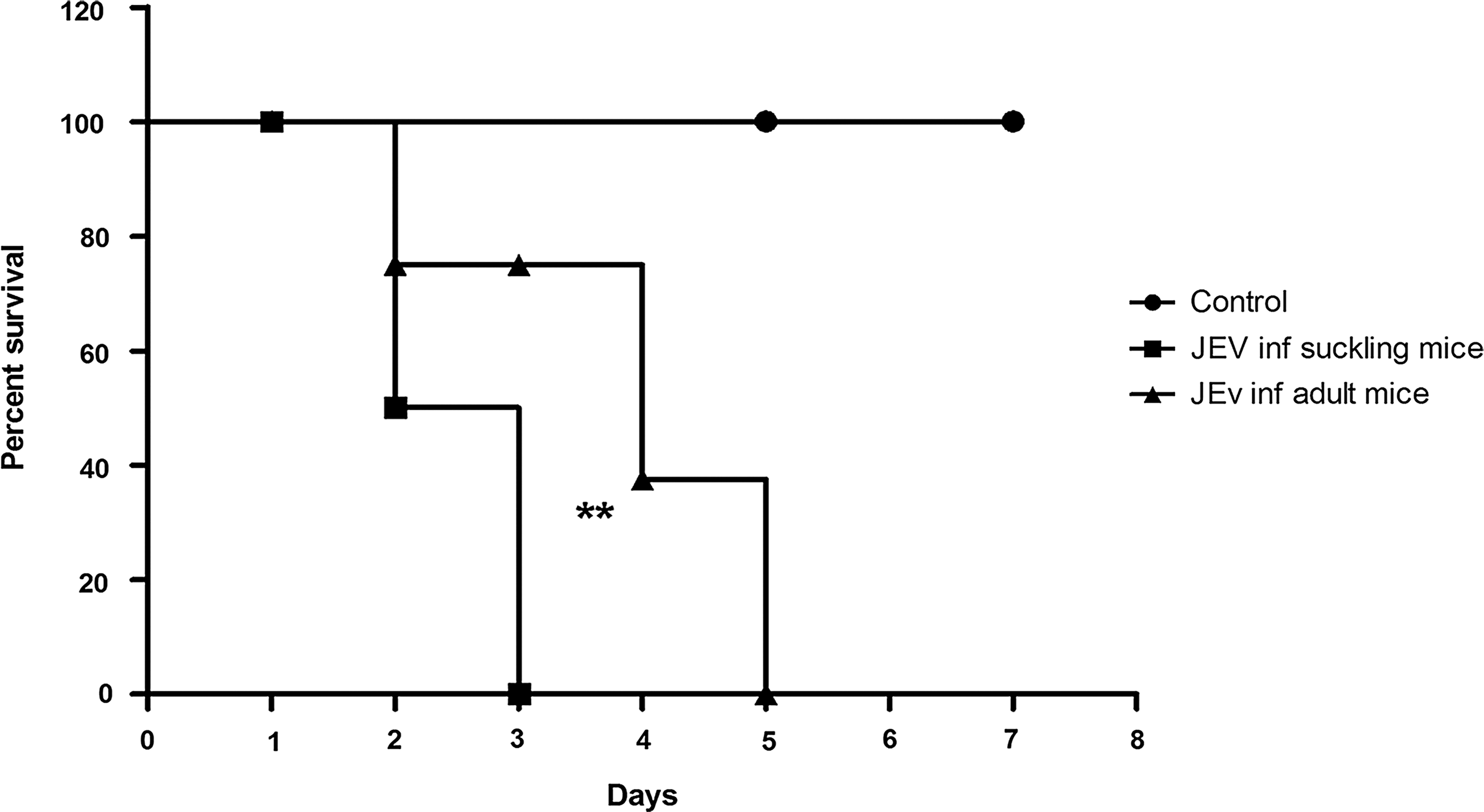
Kaplan Meyer plots depicting survival of JEV infected suckling and adult mice as compared to mock infected control mice. Note that death was delayed by 2 days in JEV-infected adult mice as compared to JEV-infected suckling mice. The difference between the two curves is highly significant (**) by Log-Rank test (p=0.0092).
Expression of PRRs in suckling and adult mouse brains
Brain tissue from mock-infected suckling and adult mice showed expression of all TLR genes except TLR3. However, TLR3 expression was noted in JEV-infected brain tissue. (Fig. 1, Panel B and C). RIG-I and MDA5 gene expression was not detected in infected or uninfected mouse brains by conventional PCR.
As evident from Figure 2, mRNA obtained from brain tissue of suckling mice infected with JEV exhibited significant upregulation of TLRs 2, 3, 4, 5, 6, and 8. On the other hand, RIG-I expression was seen to be downregulated in JEV-infected suckling mice as compared to mock-infected mice. In adult mice, TLR2, 3, 4, 7, and 9 showed a significant increase compared with mock-infected mice, while TLR8 and RIG-I were downregulated. It was noted that TLR2 expression increased drastically in JEV-infected adult mice (103-fold) compared with mock-infected and JEV-infected suckling mice (3.3-fold increase). The only discrepancy in TLR gene expression between JEV-infected adult mice and suckling mice was seen for TLR8. TLR8 was upregulated in suckling mice, whereas it was downregulated in adult mice. It was observed that MDA5 was 29-fold upregulated in JEV infected adult mice, whilst MDA5 showed only 1.81-fold upregulation in suckling mice as compared to their respective mock-infected mice, although such an upregulation was not evident in conventional PCR. The probable difference between the two techniques may be related to the sensitivities of the two assays or the use of different set of primers in these assays.
Expression of PRR cascade pathway genes
Cascade genes in TLR and RIG-I like receptors (RLRs) pathways were also studied by real-time PCR (Fig. 2). Suckling mice showed Myd88 downregulation after JEV infection, while IRF3, 7, IFNα and β, TNFα, NFκB, and IL12 gene expressions were upregulated as compared to mock -infected mice. On the other hand, adult mice exhibited a different pattern of cascade pathway gene expression; upregulation of Myd88, IRF7, inteferon α, TNFα, NFκB, and IL12 as compared to mock-infected mice. In addition, adult mice infected with JEV showed downregulation of IRF3, and IFNβ as compared to mock infected mice (Fig. 2).
PCR array findings for TLR signaling
Several Myd88-independent pathway genes were upregulated (IRF3, TRIF, TRAF6 etc.) in JEV-infected Neuro2a cells and the brains of suckling mice (Fig. 4, Panel B) as compared to the Myd88-dependent pathway. On the contrary, in the brains of JEV-infected adult mice (Fig. 4, Panel A), a number of Myd88-dependent pathway genes (TLR2, TLR9, Myd88) were upregulated.
The expression of several cytokine regulatory genes (IL6, CCL2, TNFα, IL12) were grossly upregulated following JEV infection in Neuro2a cells and adult brains but not in the brains of suckling mice (Fig. 4, Panel C). Differential expression was also noted between JEV-infected adult and suckling mice with respect to certain other adaptive immune system related genes such as CD80, CD86, IL10, and IL2 (Fig. 4, Panel D).
Interferon regulatory genes (INFγ, INFβ, and CXCL2) were upregulated in JEV-infected Neuro2a, suckling, and adult brain cells. However, the degree of upregulation varied with adult mice brain cells (Fig. 4, Panel E) exhibiting a fold change of 122, 106, and 204, respectively. The fold change observed in Neuro2a cells was 59.8, 40.22, and 87.2, respectively. Interestingly, the fold change in these three genes was less than 50 in suckling mice brain cells.
The effector genes that are related to apoptosis and cell death such as FADD, CASP8, and PPARA were upregulated more in JEV-infected Neuro2a cells (58.8, 5, 28-fold) and suckling mice brain (38, 20, 23-fold), respectively, while they were either marginally upregulated (PRKR, TRAF6, and CASP8) or downregulated (PPARA and FADD) in brain tissue of JEV-infected adult mice (Fig. 4, Panel F). A full list of the changes in gene expression in these experiments is presented in Table 2.
Confirmation of role of TLR3 in JEV-infected Neuro2a cells
The silencing of the TLR3 gene in Neuro2a cells was confirmed by comparing the Ct values in JEV-infected and JEV-infected TLR3 gene silenced Neuro2a cells. TLR3 gene was upregulated (4.85-fold) in JEV-infected Neuro2a cells (Table 3). On the contrary, the downregulation of TLR3 gene was −1.38 fold in JEV-infected TLR3 silenced Neuro2a cells (Table 3). This difference was highly significant (p<0.05). This was further substantiated by the observation that the Ct values for JEV NS5 gene were far lower (16.67) in JEV-infected TLR3 gene silenced Neuro2a cells as compared to JEV-infected Neuro2a cells (25.50), thereby indicating enhanced virus production (>2 logs) in TLR3 gene silenced cells (Table 3 and Fig. 6).
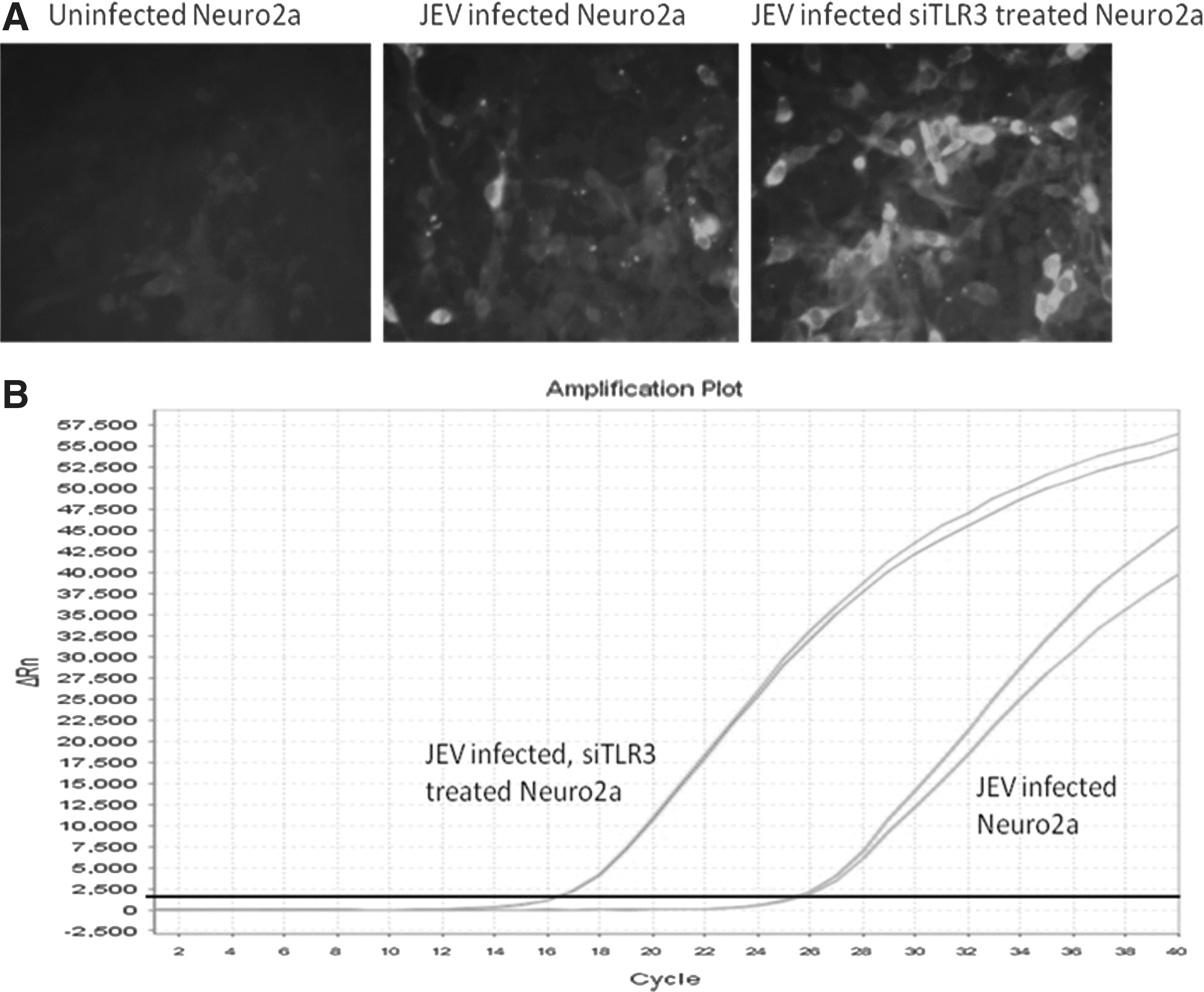
Confirmation of TLR3 in JEV infection in Neuro2a cell line at 18 h post infection.
JEV-NS5 gene was used to estimate viral RNA in infected Neuro2a cells. The mean Ct values for JEV-infected Neuro2a was 25.5 and for JEV-infected TLR3 silenced Neuro2a cells was 16.67.
Discussion
Significant strides have been made in understanding the innate immune response mediated by TLRs following infection with different viruses. However, not all events following TLR activation have been fully understood. The outcome of a virus–TLR interaction is complex and depends upon the particular TLR and the virus in question, as well as host species (2,4,5). There are very few studies that have addressed this issue in the context of JEV. Recently, Gupta et al. (13) studied the global transcription profile in the brains of mice infected with JEV and reported that TLRs 2, 3, and 7 were upregulated. The present study also revealed that these three TLRs are upregulated following JEV infection in suckling mice. Nazmi et al. (17) reported that JEV infection results in increased expression of RIG-I in the neurons of mice. Further, they noted that virus infection resulted in activation of RIG-I leading to modulation in the downstream pathways that cause the infected neurons to participate in the inflammatory milieu in the CNS, which ultimately leads to their own demise (18). On the contrary, the observations of the present study did not reveal any change in RIG-I expression in JEV-infected neurons. This may be attributed to the differences in the study design. Nazmi et al. (2011) used GP78 strain of JEV, BALB/c mice and an MOI of 5, while in the present study, P20778 strain of JEV, C57BL6/J mice and an MOI of 0.1 were used. Although these studies (13,17,18) have added new information and thrown some light on the role of a few PRRs in JEV infection, they were not designed to comprehensively address the gamut of innate immune events that occur following JEV infection, especially delineating the role of all the PRRs. Furthermore, the cascade events that result following JEV infection in vitro as well as in vivo have not been identified. Therefore, this study was designed to investigate the role of all PRRs in JEV infection with special emphasis on understanding the role of TLRs and other PRRs in JEV infection in vitro using mouse neuronal cell line and compare with events that occur in vivo using JEV-infected suckling and adult mice.
In an initial screening assay, using conventional PCR and commercially available primers for all TLRs 1–9, RIG-I, and MDA5 genes, it was observed that TLR3 gene expression was prominent in JEV-infected Neuro2a cells while it was absent in uninfected cells (Fig. 1, Panel A). An identical observation was also made in vivo wherein JEV infected mouse brain tissue obtained from both adult and sucking mice showed differential expression of TLR3 gene as compared to mock infected animals (Fig. 1, Panel B and C). None of the other TLR genes (TLRs 1, 2, and TLRs 4–10) or other PRR genes (RIG-I and MDA5) exhibited such a differential expression either in vitro in Neuro2a cells or in vivo in the mouse brain tissue. In order to validate these observations, real-time PCR amplification was carried out. The real-time PCR for PRRs revealed a differential upregulation of TLR3 gene, as well as few other genes (TLR1, 2, and 8) both in Neuro2a cells and mice. TLR3 was upregulated 5.8-fold and 7.8-fold in JEV-infected mouse brain tissue of suckling mice and adults, respectively (Fig. 2).
In addition, PCR array results of JEV-infected Neuro2a cells, as well as suckling mice and adult mice brains, revealed that the signaling pathway related genes such as IRF3, TRAF6, NFκB, and IFNβ were upregulated (Fig. 4). These observations suggest that TLR3 receptors were triggered following JEV infection both in vitro and in vivo, resulting in a cascade of events including upregulation of cytokine regulatory genes (Il6, TNFα, and CCL2) and effector regulatory genes (FADD, PPARK, and CASP8), leading to a strong pro-inflammatory response, resulting in apoptotic cell death. TLR3 has been implicated in the pathogenesis of West Nile virus infection, wherein it was reported that it contributed to lethality of mice by promoting peripheral inflammation that led to the breakdown of the blood-brain barrier, resulting in increased viral load in the brain of infected mice (21). In order to ascertain whether TLR3 is indeed a key innate immune molecule modulating JEV infection in neurons, we adopted a TLR3 gene silencing approach. The results presented here unambiguously demonstrate that silencing of TLR3 gene was associated with significant enhancement of JEV replication in Neuro2a cells (Table 3, Fig. 6). Such a phenomenon has not been reported earlier in JEV infection, although Daffis et al. (2008) have demonstrated that TLR3 serves a protective role against WNV in part by restricting replication in neurons. However, further research is required to confirm whether the TLR3 engagement in JEV infection is also operative in human patients with JE.
The two other significant observations in this study were gross upregulation of TLR2 and MDA5 noted in JEV-infected adult mice but not in suckling mice or Neuro2a cells infected with JEV (Fig. 2). TLR2 has traditionally been implicated in the recognition of bacterial lipoproteins/peptidoglycans (19,20). However, involvement of TLR2 in a number of viral infections such as herpes simplex virus 1, Epstein Barr virus, vaccinia virus, lymphocytic choriomeningitis virus, respiratory syncytial virus, and hepatitis C virus has been reported earlier (4,5). In all these viral infections, it has been suggested that TLR2 plays a crucial role in mounting a potent antiviral response. Among the JEV infection models investigated in this study, TLR2 genes were upregulated more than 100 fold in brain tissue of adult mice infected with JEV as compared to control uninfected mice. In contrast, suckling mice infected with JEV by the intracerebral route and Neuro2a cells infected with JEV in vitro did not show such a marked increase (Fig. 2). This observation, coupled with the fact that the onset of symptoms and death were delayed in adult mice (Fig. 5), suggests that TLR2 gene is triggered in JEV infection in adult mice followed by a cascade of events (Fig. 4), resulting in increased expression of Myd88 (3.4 fold) and IRF7 (34 fold), TNFα (2.5 fold), and IL12 (12.8 fold), all of which have been implicated in mounting an antiviral response. In addition, the PCR array data has also revealed upregulation of interferon and chemoattractant genes (IFNγ, IFNβ, and CXCL10), as well as adaptive immune regulatory genes (CD80 and CD86) in the brains of adult mice infected with JEV (Fig. 4).Taken together, all these observations therefore suggest that in adult mice there is a robust attempt made by the innate immune system to defend against JEV infection by using an unconventional TLR2 route that could have resulted in delayed onset of symptoms and death.
MDA5 was also significantly upregulated (29 fold) in JEV-infected adult mice, but not JEV-infected suckling mice or Neuro2a cells in vitro. The only report of involvement of MDA5 in JEV has been in porcine kidney (PS) cells (9). It is tempting to postulate that the MDA5 upregulation noted only in adult mice is probably an evasive strategy adopted by the virus to counter the antiviral response mediated through engagement of TLR2 receptor. Though there was TLR2 upregulation in JEV-infected adult mice, the resultant end products of TLR2 activation did not reveal significant upregulation of interferons. This probably suggests that the virus evaded the host antiviral TLR2 response. The increased expression of MDA5 correlated with an absence of the downstream product of TLR2 signaling, that is type I interferon production. Thus, the virus was able to overcome the innate immune protective mechanisms resulting in death of mice.
Although the expression levels of other PRRs were different in Neuro2a, suckling and adult mice brain, it can be explained by the fact that the maturation level of neuronal cells in these three groups are different; Neuro2a cells and neurons of suckling mouse brain are relatively ‘immature’ or at a different maturation level as compared to neurons derived from adult mouse brain.
In conclusion, this is the first report to have addressed the role of innate immune receptors in JEV infection comprehensively. The novel findings in this study include the key role of TLR3 in mediating a protective response and delineation of the role of MDA5 as supportive of viral replication deleterious to the host. Further evaluating the role of TLR2 in early protection of the host needs to be addressed in humans with JE.
Footnotes
Acknowledgment
PRF was supported by the UK Medical Research Council grant: INDO-UK-JE/VR002. LT is supported by Wellcome Trust MB PhD Postdoctoral Fellowship no. 087757/Z/08/Z.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.