
Abstract
We recently found that the herpes simplex virus-1 (HSV-1) latency-associated transcript (LAT) results in exhaustion of virus-specific CD8+ T cells in latently-infected trigeminal ganglia (TG). In this study we sought to determine if this impairment may involve LAT directly and/or indirectly interfering with DC maturation. We found that a small number of HSV-1 antigen-positive DCs are present in the TG of latently-infected CD11c/eYFP mice; however, this does not imply that these DCs are acutely or latently infected. Some CD8+ T cells are adjacent to DCs, suggesting possible interactions. It has previously been shown that wild-type HSV-1 interferes with DC maturation. Here we show for the first time that this is associated with LAT expression, since compared to LAT(−) virus: (1) LAT(+) virus interfered with expression of MHC class I and the co-stimulatory molecules CD80 and CD86 on the surface of DCs; (2) LAT(+) virus impaired DC production of the proinflammatory cytokines IL-6, IL-12, and TNF-α; and (3) DCs infected in vitro with LAT(+) virus had significantly reduced the ability to stimulate HSV-specific CD8+ T cells. While a similar number of DCs was found in LAT(+) and LAT(−) latently-infected TG of CD11c/eYFP transgenic mice, more HSV-1 Ag-positive DCs and more exhausted CD8 T cells were seen with LAT(+) virus. Consistent with these findings, HSV-specific cytotoxic CD8+ T cells in the TG of mice latently-infected with LAT(+) virus produced less IFN-γ and TNF-α than those from TG of LAT(−)-infected mice. Together, these results suggest a novel immune-evasion mechanism whereby the HSV-1 LAT increases the number of HSV-1 Ag-positive DCs in latently-infected TG, and interferes with DC phenotypic and functional maturation. The effect of LAT on TG-resident DCs may contribute to the reduced function of HSV-specific CD8+ T cells in the TG of mice latently infected with LAT(+) virus.
Introduction
DCs are powerful sentinels in innate and adaptive immunity, due to their unique and critical role in priming (initial activation) of naïve T cells and recall of antiviral memory T-cell responses (21
–27). The maturation status of DCs determines whether they induce or tolerize CD8+ T cells (28). DCs would therefore be the most crucial and potent antigen-presenting cells (APC) in presenting CD8+ T-cell epitopes during the early course of virus reactivation to stimulate TG-resident memory CD8+ T cells. CD8+ T-cell numbers and their ability to reduce virus replication in the TG are increased in CD11c−/− deficient mice (29). It is well known that in vitro HSV-1 can efficiently infect DCs in their immature state, and interfere with their phenotypic maturation (3,26,30
–32). This could represent an immune-evasion mechanism that helps HSV-1 escape the host's CD8+ T-cell immune surveillance. However, the HSV genes involved in downregulating DC functions are unknown. As LAT is the only HSV-1 gene abundantly transcribed during latency, it is a candidate for modulating DC Ag-presenting function during latent infection (33
–35). We hypothesize that LAT inhibits phenotypic maturation of DCs, and subsequently impairs their function of stimulating TG-resident CD8+ T cells. This proposed LAT function might be one of the mechanisms involved in increased reactivation of LAT(
The results reported here strongly suggest that in vitro the HSV-1 LAT: (1) reduces DC proinflammatory cytokine production; (2) interferes with cell surface expression of MHC-I and the co-stimulatory molecules CD80 and CD86; and (3) reduces the ability of HSV-1-infected immature DCs to stimulate HSV-specific CD8+ T cells. In vivo, the data show that mouse TG latently infected with LAT(
Materials and Methods
CD11c/eYFP transgenic mice
CD11c/eYFP transgenic mice originally developed by Dr. Michel C Nussensweig (Rockefeller University) were kindly provided to us by Dr. James T. Rosenbaum (Oregon Health & Science University). CD11c/eYFP transgenic mice have been successfully used for in vivo tracking of activated DCs (36,37). C57BL/6 (B6) mice, 5 wk old, were purchased from the Jackson Laboratory (Bar Harbor, ME). The animal studies conformed to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH). The mice were maintained in air-filtered cages and fed normal mouse chow in the University of California–Irvine (UCI) Animal Care Facility. The UCI Committee on Animal Research approved all procedures (ID #2002-2372).
Virus titration
Plaque-purified HSV-1 strain McKrae (wild-type), and its derived mutants dLAT2903 LAT(−) and dLAT2903R LAT(+) were grown in rabbit skin (RS) cell monolayers in minimal essential medium (MEM) containing 5% fetal calf serum (FCS), as described previously (20).
Ocular infection
Mice were infected ocularly without scarification with 2×105 pfu of either LAT(+) or LAT(−) virus. All viruses were suspended in 5 μL of tissue culture media and administered as an eye drop. Mock-infected mice received sterile tissue culture media.
Preparation of single-cell suspensions from mouse TG
Mice were euthanized and TG were harvested, pooled, and digested in DMEM-5% FBS containing collagenase I (Life Technologies, Carlsbad, CA), as we previously described (5 –8,38). The digested tissue suspension was passed through a 45-mm nylon cell strainer. Cell suspensions were spun down at 1400 rpm for 5 min at 4°C, washed, and suspended in FACS buffer (PBS-0.01%, NaN3-0.1%, BSA, and 2 mM EDTA) for FACS acquisition and analysis. Ten TG were pooled and digested with collagenase type I. To obtain the number of cells per TG, the total numbers of cells obtained from each 10-TG cell suspension were divided by 10. The cells were stained with mAbs and tetramers, as described below. The lymphocyte populations in the TG suspension were gated from other non-lymphocyte cells (i.e., glial cells, Schwann cells, neurons, and fibroblasts) based on their specific size and granularity. The total number of CD8+ T cells was calculated based on the number of gated lymphocytes and used for normalization.
Tetramer assay
TG cell suspensions were analyzed for the frequency of CD8+ T cells specific to the HSV-gB498–505 epitope using the HSV-gB498–505 tetramer, which is H2-Kb-restricted, as we previously described (39). The cells were first incubated with 2 μL of PE-labeled HSV-gB498–505/H2-Kb tetramer at 4°C for 30–45 min. The cells were washed twice and stained with 1 μL FITC-conjugated rat anti-mouse CD8 antibody (clone 53-6.7; eBioscience, San Diego, CA). After two additional washings, the cells were fixed with 1% formaldehyde in PBS. A total of 400,000 events were acquired by FACScan (Becton Dickinson, San Jose, CA), followed by analysis using Cell Quest software (BD Biosciences, San Jose, CA). The absolute numbers of HSV-gB498–505-specific CD8+ T cells were calculated using the following formula: (Nbr of CD8+ tetramer+ cells in the test) − (Nbr of CD8+ tetramer+ cells in the negative control). The anti-mouse CD8 mAb was pre-tested prior to use to confirm that it did not interfere with or alter tetramer binding.
CD107 cytotoxicity assay
The CD107 assay was performed as recently described (5 –10,40), with a few modifications. Freshly isolated TG-derived cells were left unstimulated, or were stimulated with gB498–505 or PHA (positive control) at 37°C for 5–6 h in the presence of BD GolgiStop (BD Biosciences) and 10 μL of FITC-conjugated CD107 antibody. The cells were washed and stained with 2 μL of PE-labeled HSV-gB498–505/H2-Kb tetramer, and 1 μL of PerCP-conjugated anti-mouse CD8 for 30 min at 4°C. The cells were then washed again and analyzed using a FACScan (BD Biosciences).
Cytokine assay
Dendritic cells were cultured in 6-well plates at 5×106 cells/well in CM infected with HSV-1 at MOI 0.3, or incubated with LPS at 0.5 μg/mL as positive control. The supernatant was harvested from infected DCs every 6 h for 36 h after infection and/or LPS stimulation, and the concentrations of IFN-γ, tumor necrosis factor-α (TNF-α), and IL-12 cytokines were determined using sandwich ELISA kits specific for each cytokine, according to the manufacturer's instructions (BD Pharmingen, San Diego, CA).
Immunohistochemistry
Immunostaining of latently-infected TG was performed on day 35, as we previously described (5,6,7,8,20). Briefly, the mice were euthanized, and TG were harvested, embedded in embedding compound, and snap-frozen. Approximately 10-μm-thick cryosections were made, fixed in acetone (10 min at room temperature), air-dried, and stored at −80°C. For immunostaining, TG sections were rehydrated in PBS (10 min at room temperature), Fc blocked, and then stained with a rabbit antibody to HSV-1 (catalog no. F031802; Dako, Carpenteria, CA), followed by an anti-rabbit IgG-conjugated with bright orange fluorescent Alexa-Fluor 546 dye (which is spectrally similar to Cy3 dye)-labeled rat anti-HSV-1 antibody (catalog no. A-11071; Molecular Probes, Eugene, OR), at 1:100 dilution in PBS-1% BSA at 4°C overnight. After three successive washings in PBS (3×5 min), the sections were stained with 14.3 mM 4,6-diamino-2-phenylindole (DAPI; Molecular Probes) for 2 min at room temperature to stain cell nuclei. Excess DAPI was removed by washing with PBS (3×5 min), and the slides were mounted in 50% glycerol-PBS and analyzed by fluorescence microscopy.
DNA extraction and real-time PCR analysis
DNA was isolated from individual TG using the commercially available DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA), according to the manufacturer's instructions. For real-time PCR assays, 100 ng of DNA from all samples was subjected to PCR using HSV-1 gB-specific primers. The following primers were used for HSV-1 gB: forward: 5′-AACGCGACGCA CATCAAG-3′; reverse: 5′-CTGGTACGCGA TCAGAAAGC-3′. The amplicon length for this primer set is 72 bp. The primer concentration was 200 nM for both strands. SYBR green master mix (Applied Biosystems, Foster City, CA) was used in a final volume of 25 μL. Cycling parameters were 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 25 sec, and 60°C for 60 sec. The plates were read after each cycle, and a melting curve was generated after amplification using an MJ Research Opticon thermal cycler. PCR controls without reverse transcriptase (water control), or with mock-infected mouse TG genomic DNA, were routinely negative. For relative copy numbers of gB DNA in mice TG, the obtained CT was translated into the number of copies using standard curves generated from titrated HSV-1 viral DNA.
Generation of bone marrow-derived dendritic cells
Bone marrow-derived DCs were generated from B6 mice, as we previously described (41,42). Briefly, single-cell suspensions of bone marrow cells were cultured in 100-mm Petri dishes at an initial density of 2×105 cells/mL in a final volume of 10 mL RPMI-1640 medium supplemented with 50 ng/mL murine GM-CSF and 50 ng/mL IL-4 (PeproTech Inc., Rocky Hill, NJ). After 8–10 days, over 90% of the non-adherent cells had acquired typical dendritic morphology and were at an immature stage. The immature CD11c+/CD11b+ DCs express low levels of MHC, co-stimulatory, and adhesion molecules. Routinely ∼6×107 cells are obtained from one mouse. Differentiation of bone-marrow cells into DCs, which contain a mixture of both plasmacytoid DC and myeloid DC subsets (41,42), was followed by flow cytometry analysis of different surface markers.
DC-CD8+ T-cell assay
Immature DCs derived from bone marrow of C57BL/6 mice as described above were infected in vitro for 4 h with wild-type McKrae (LAT(+)) or dLAT2903 (LAT(−); MOI=3), and then stimulated for 16 additional hours with 0.5 μg/mL LPS to induce maturation. The cells were then washed and treated with mitomycin C, and the indicated increased numbers of DCs were incubated in duplicate for 72 h with a constant number (105) of autologous CD8+ T cells derived from wild-type McKrae-infected C57BL/6 mice. The number of HSV-specific CD8+ T cells producing IFN-γ was determined by an ELISpot assay, as we previously described (43).
Statistical analysis
The statistical module Prism was used to perform unpaired two-tailed Student's t-tests and analysis of variance (ANOVA) with Tukey's t-test. All error bars represent the standard errors of the means. A p value <0.05 was considered significant.
Results
More HSV-1 Ag-positive DCs in the TG of CD11c/eYFP transgenic mice latently-infected with LAT(+) virus compared to LAT(−) virus
CD11c/eYFP transgenic mice have been recently used for in vivo tracking of CNS-resident DCs (36,37). Two groups of CD11c/eYFP transgenic mice (n=10) were infected ocularly with: (1) the LAT(−) mutant dLAT2903; or (2) the parental wild-type McKrae. Thirty-five days post-infection (when latency was fully established), the TG were harvested, and (1) the presence of TG-resident HSV Ag-positive CD11c+ DCs was detected by confocal microscopy, (2) the number of DCs per TG was determined by FACS, and (3) the amount of viral DNA was determined by qRT-PCR. A small number of HSV-antigen-positive DCs were detected in the TG of CD11c/eYFP transgenic mice latently-infected with LAT(+) HSV-1 (Fig. 1A). Some CD11c+ DCs appeared to be co-localized or adjacent to CD8+ cells in the TG of both LAT(+)- (Fig. 1B) and LAT(−)-virus infected mice, suggesting that an in vivo DC-CD8 interaction may be occurring in latently-infected TG. Similar numbers of CD11c+ DCs were detected in the TG of CD11c/eYFP transgenic mice 35 d post-infection with either LAT(+) or LAT(−) virus (Fig. 1C; FACS). The bar graph in Fig. 1D shows that the average number of HSV-1 antigen-positive DCs detected by FACS in a pool of LAT(+) TG was 15, compared to just 6 in LAT(−) TG. Consistent with previous studies (44,45), more HSV-1 DNA was detected in LAT(+)-infected TG compared to LAT(−)-infected TG (Fig. 1E).
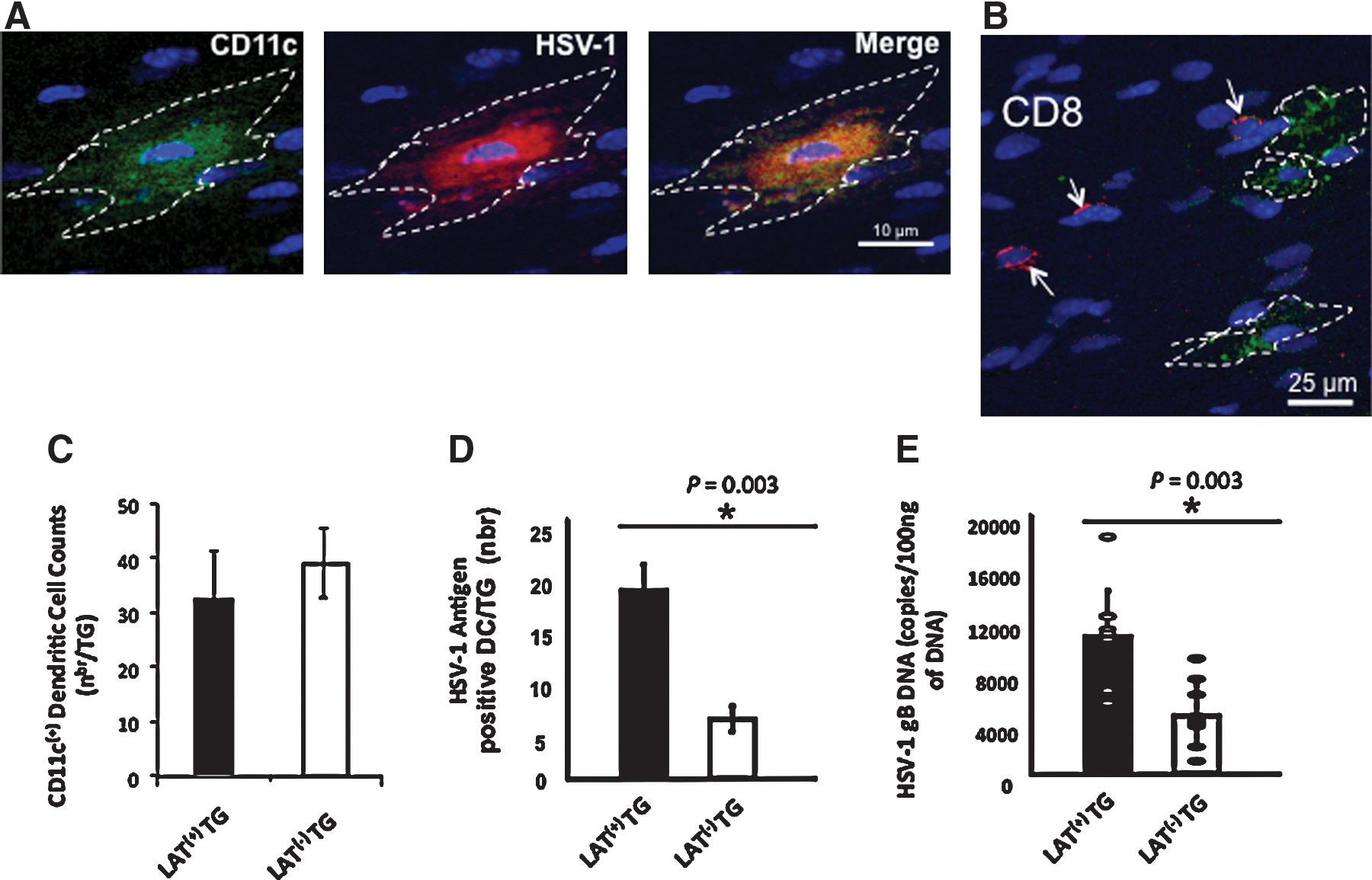
A small number of HSV-Ag-positive dendritic cells are detected in the TG of CD11c/eYFP transgenic mice latently infected with LAT(+) virus. TG from CD11c/eYFP transgenic mice latently infected with either LAT(+) (wild-type McKrae), or LAT(−) null mutant (dLAT2903) virus were removed 35 d post-ocular infection, fixed, embedded in embedding compound, immunostained as indicated, and examined by confocal microscopy, as described in the materials and methods section. All images were captured as grey scale and pseudocolored. YFP channel is shown as green. PE and Cy5 channels are shown as red. DAPI is blue. (
Because of the low number of DCs/TG (an average of 15 DCs/LAT(
LAT interferes with phenotypic maturation of DC
Immature DCs in vitro were left uninfected or infected with either LAT(+) (dLAT2903R) or LAT(−) (dLAT2903) virus. Four hours post-infection, each DC population was stimulated with LPS for maturation, and then examined by flow cytometry for the DC maturation markers MHC class I (MHC-I), and the co-stimulatory molecules CD80 and CD86. CD40 adhesion molecule was included as a control. LPS stimulated LAT(−)-infected DCs induced significant maturation of DCs, with significant increases of cell surface expression of MHC-I, CD80, and CD86 co-stimulatory molecules, compared to LAT(+)-infected immature DCs (Fig. 2A, left panels; p<0.005). LPS stimulation did not affect the surface expression of other DC markers that are unrelated to DC maturation, such as CD40 (or CD11c and CD54; not shown), suggesting that the changes in surface expression of the DC maturation markers was not due to global changes in cell surface expression. Fig. 2B shows the average mean fluorescent intensities (MFI), with each bar representing the average±SD of four independent experiments. Together these results indicate that DC phenotypic maturation was impaired by LAT(+) virus.
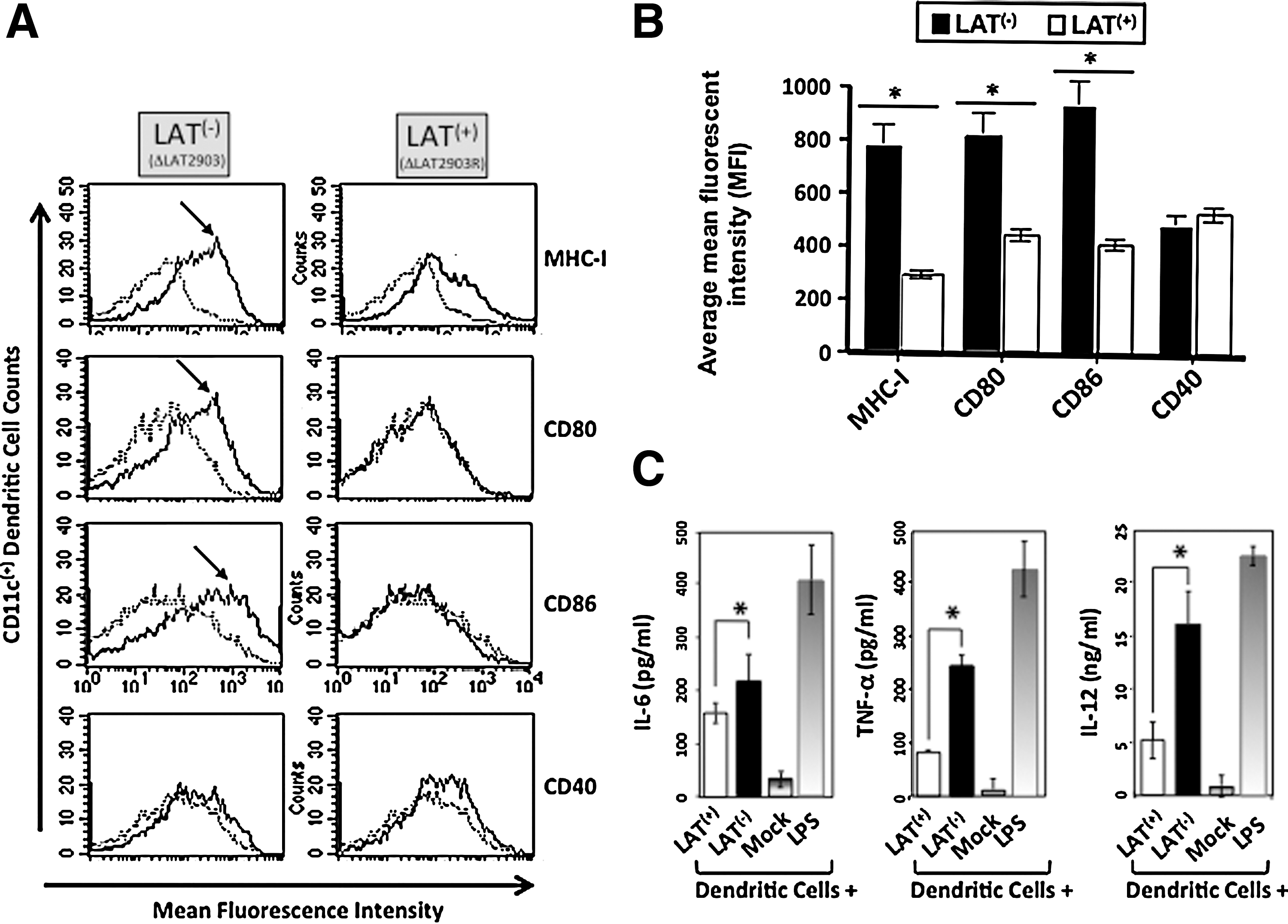
Immature DCs infected with LAT(+) virus had impaired phenotypic maturation and production of proinflammatory cytokines. (
Next, we determined whether in vitro infection of immature DCs with LAT(+) virus has an effect on the production of proinflammatory cytokines. Four hours after infection of immature DCs with either LAT(+) or LAT(−) viruses, they were subjected to LPS stimulation. Negative and positive controls were mock-infected (mock), and LPS-matured DCs, respectively. Production of IL-6, TNF-α, and IL-12 were quantified by sandwich ELISA, as described in the materials and methods section. DCs infected with either LAT(−) or LAT(+) viruses produced more IL-6, TNF-α, and IL-12 than mock-infected DCs (Fig. 2C; p<0.005). However, LAT(+)-infected DCs produced significantly less IL-6, TNF-α, and IL-12 than did LAT(−)-infected DCs (Fig. 2C; p<0.005). As expected, the largest amount of proinflammatory cytokines were detected in uninfected LPS-matured DCs. Thus, compared to LAT(−) HSV-1-infected DCs, LAT(+) HSV-1-infected DCs had decreased production of proinflammatory cytokines following stimulation with LPS.
LAT interferes with functional maturation of DCs
We further explored whether the LAT(+)-mediated inhibition of phenotypic maturation of DCs would translate into a functional impairment in their ability to induce HSV-specific CD8+ T cells. Immature bone marrow DCs were derived from H2b mice, and infected for 4 h with LAT(−) or LAT(+) virus, and then incubated with LPS for an additional 16 h, as described above, to induce their maturation. Increasing numbers of infected DCs were added to a constant number (105) of CD8+ T cells isolated from the spleens of HSV-1-infected H2b mice 35 days post-infection. LAT(−)-infected DCs induced significantly more IFN-γ-producing CD8+ T cells than LAT(+)-infected DCs (p<0.05; (Fig. 3), and this occurred in a dose-dependent manner.
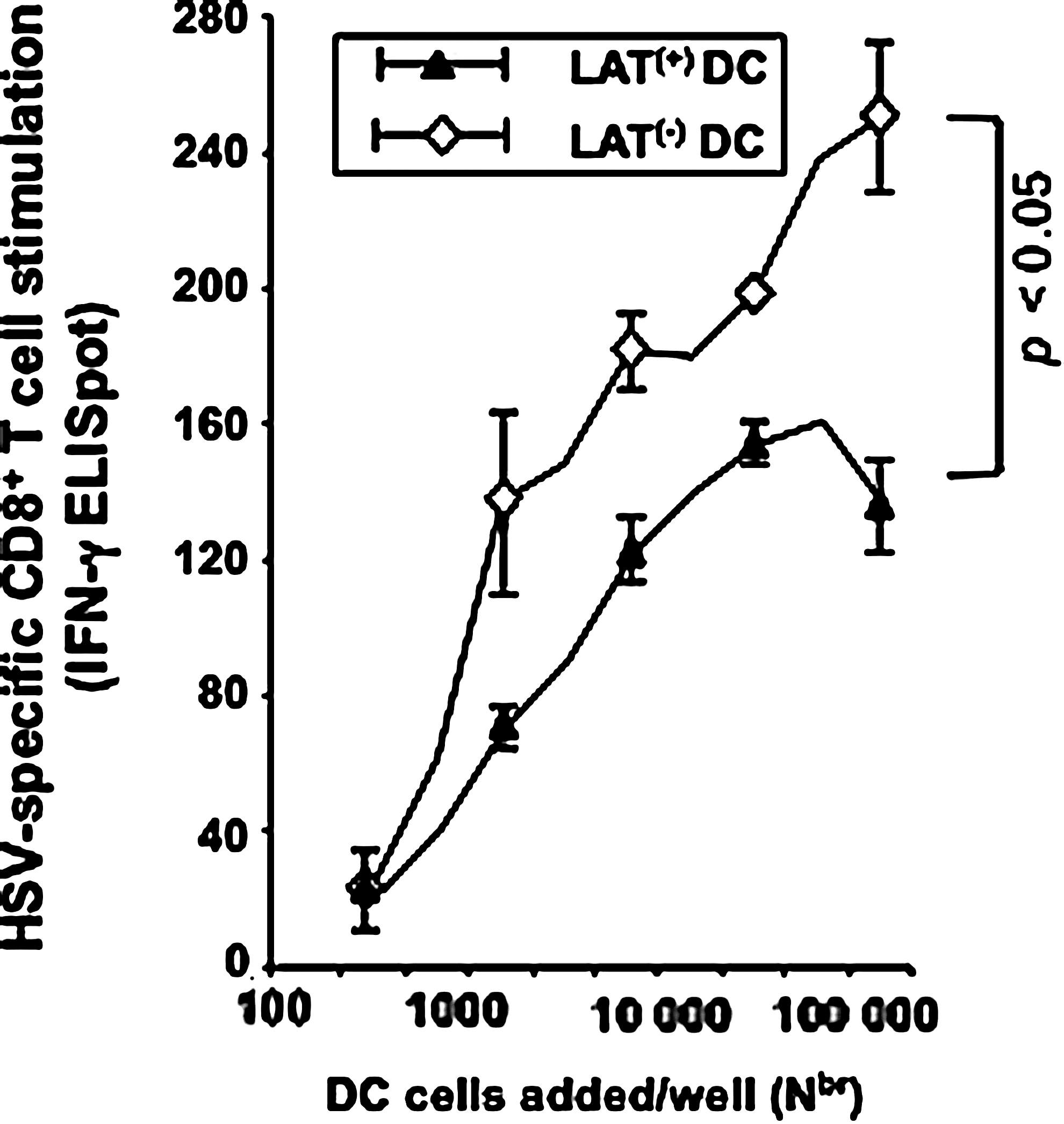
Compared to DCs infected with LAT(−), immature DCs infected with LAT(+) virus are inefficiently matured by LPS (as judged by their poor ability to stimulate CD8+ T cells). Immature bone marrow-derived DCs were infected in vitro for 4 h with LAT(+) or LAT(−) HSV-1, and incubated for 16 additional hours with 0.5 μg/mL LPS to induce maturation. The DCs were washed, and treated with mitomycin C, and the indicated numbers of DCs were incubated in duplicate for 72 h with 105 autologous CD8+ T cells derived from spleens of wild-type McKrae-infected mice. The number of HSV-specific CD8+ T cells producing IFN-γ was determined by an ELISpot assay. Results show the average±SD of two independent experiments.
Latently-infected LAT(+) TG have more total CD8+ T cells than LAT(−) TG
In a pilot experiment, we compared the percentages and total numbers of T cells in mouse TG latently infected with wild-type HSV-1 (strain McKrae), that had been perfused or not perfused prior to euthanasia. No difference was found in the percentages of total CD4+ and CD8+ T cells when we compared perfused versus non-perfused mice. This was expected, since during HSV-1 latency there are few if any HSV-1-specific T cells in the circulating blood. Subsequent experiments were carried out using non-perfused mice.
We examined whether HSV-1 LAT can affect the percentages and total number of TG-resident CD4+ and CD8+ T cells. Two groups of B6 mice (n=10) were infected ocularly with 2×105 pfu/eye with: (1) LAT(−) virus mutant dLAT2903 (LAT(−)); or with (2) its marker-rescued virus dLAT2903R (LAT(+)) (20) (Fig. 4A). Both viruses are derived from HSV-1 strain McKrae. A third group of control mice received vehicle alone (mock-infected). We harvested TG 35 days post-infection (i.e., during latent infection), and determined the number and the percentage of CD3+ T cells, and of CD3+CD4+ and CD3+CD8+ T-cell populations in LAT(+)- and LAT(−)-infected TG. A significant increase in the number of CD3+ T cells was detected in both LAT(+) TG and LAT(−) TG, compared to mock-infected TG (Fig. 4B, left panels). In addition, the percentages of both CD3+CD4+ and CD3+CD8+ T-cell sub-populations were increased in latently-infected LAT(+) TG and LAT(−) TG compared to mock-infected TG (Fig. 4B, right panels). Interestingly, in five consecutive independent experiments (each used 10 latently-infected mice per group), the percentages of CD8+ T cells were significantly higher in LAT(+) TG compared to LAT(−) TG (Fig. 4B, right panels). Latently-infected LAT(+) TG contained an approximately threefold higher percentage of CD3+CD8+ T cells compared to LAT(−)-infected TG. In contrast, the percentages of CD4+ T cells were similar in LAT(+) TG and LAT(−) TG (Fig. 4B, right panels). Similar results were obtained when comparing TG-resident T cells from the LAT(−) virus mutant dLAT2903 (LAT(−)) versus WT virus McKrae (LAT(+)) (not shown). We previously reported that TG from mice latently infected with LAT(+) HSV-1 contain more total CD8+ T cells than TG from mice latently infected with LAT(−) HSV-1 (47,48). The same results were obtained here. In addition, we now show that in contrast to CD8+ T cells, the total number of CD4+ T cells did not appear to vary between TG from mice latently infected with LAT(+) versus LAT(−) HSV-1. The average±SD/TG of 5 independent experiments, each consisting of cells pooled from 20 TG, is shown in Fig. 4C (left panels). Based on this result, we focused on assessing the effect of LAT on the number of CD8+ T cells in TG.
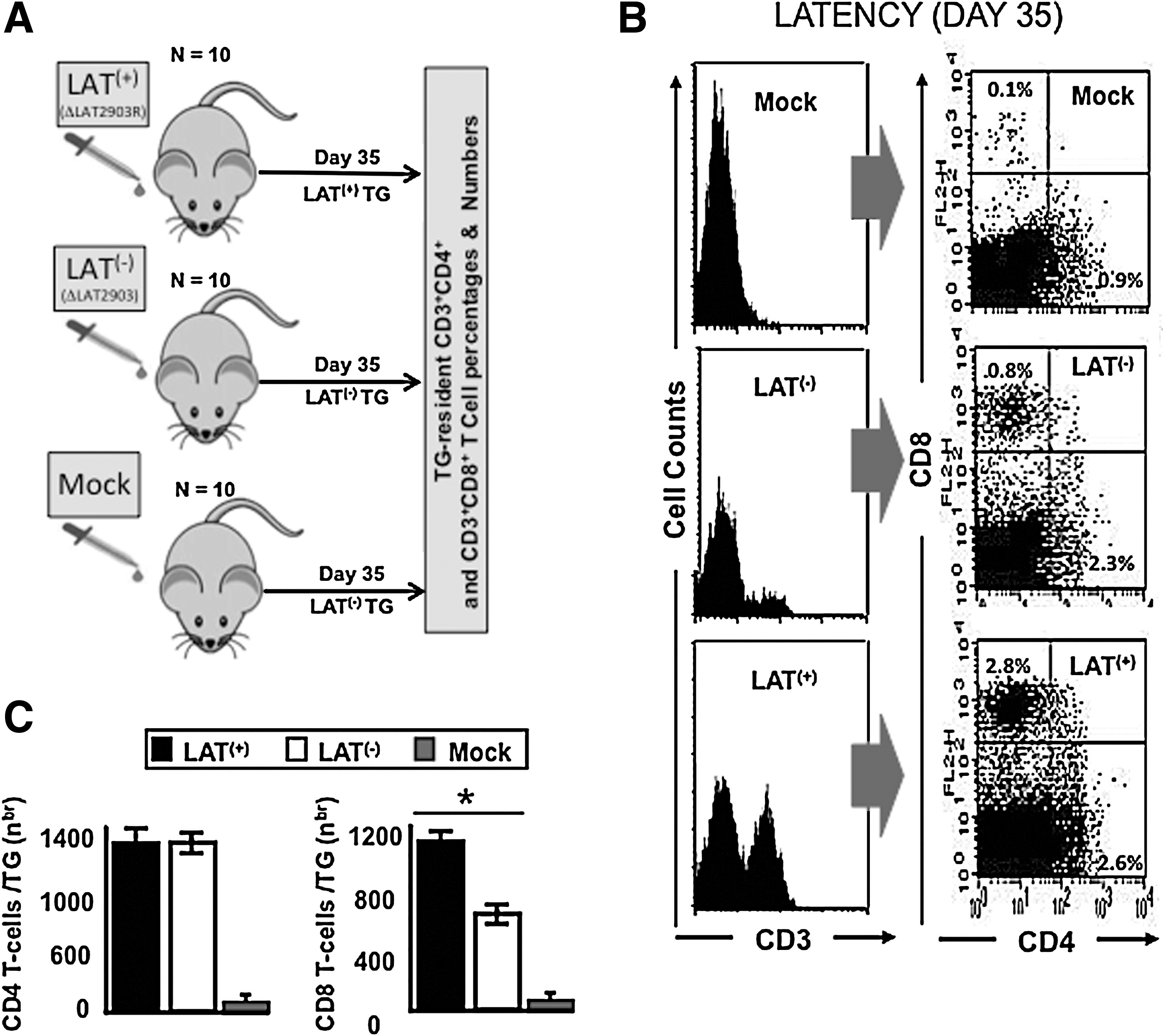
Higher percentages of total CD8+ T cells were detected in LAT(+) TG than in LAT(−) TG during latent herpes infection. (
Higher numbers of dysfunctional HSV-gB498–505-specific CD8+ T cells are present during latency in LAT(+) TG compared to LAT(−) TG
To assess the effect of LAT on the DC's ability to induce TG-resident CD8+ T-cell function in vivo, groups of 40 age- and sex-matched B6 mice were ocularly infected with either LAT(+) or LAT(−) virus. The HSV-gB498–505-epitope and B6 mice were chosen to detect HSV-1-specific CD8+ T cells in this study, because in this mouse strain the majority (over 50%) of CD8+ T cells in the periphery, as well as in latently-infected TG, are directed to this single immunodominant epitope (49 –51). TG from 10 LAT(+) and 10 LAT(−) mice were collected 35 days post-infection (i.e., during latent infection), and TG-resident CD8+ T cells were isolated (Fig. 5A). The cytotoxic activity (Fig. 5B), and cytokine production (Fig. 5C), of TG-resident CD8+ T cells were determined following in vitro stimulation with DCs pulsed with HSV-1 gB498–505 peptide, or DCs infected with wild-type McKrae. The HSV-1-specific CD8+ T-cell cytotoxic activity as determined by CD107 expression, was significantly higher in CD8+ T cells from TG of LAT(−) latently-infected mice compared to LAT(+) latently-infected mice (Fig. 5B; p=0.001). Production of IFN-γ, TNF-α, and IL-12 were also higher in CD8+ T cells derived from mouse TG latently infected with LAT(−) virus compared to LAT(+) virus (Fig. 5C). These results suggest that LAT directly or indirectly interferes with DC maturation, which then leads to exhaustion of HSV-specific CD8+ T cells.
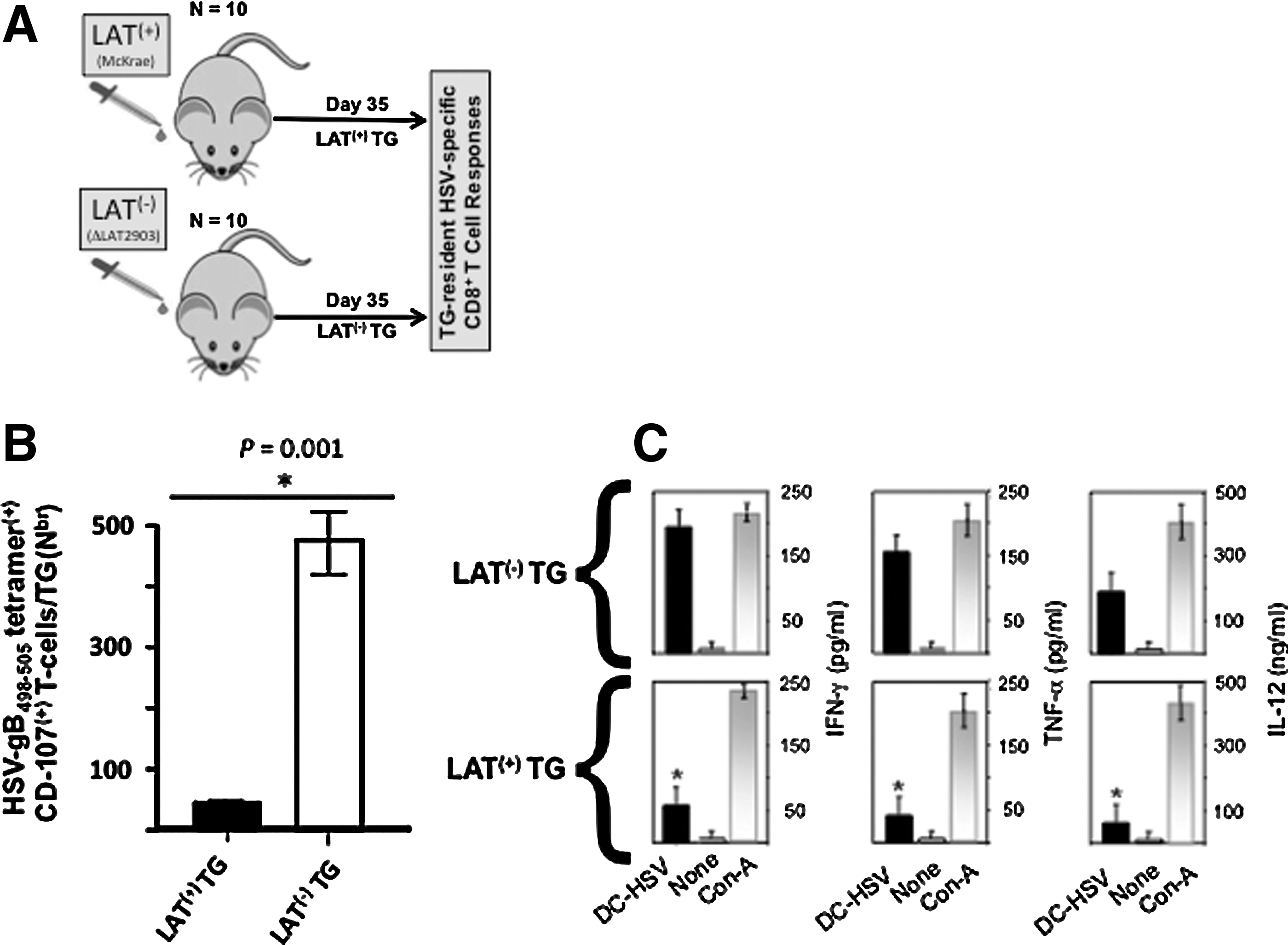
Impairment of HSV-1-specific CD8 T-cell functions in TG of mice latently infected with LAT(+) virus. (
Discussion
This report extends our recent findings regarding the role of HSV-1 LAT in inducing CD8+ T-cell exhaustion in mouse TG latently infected with HSV-1 (1,2). We show here that exposure of immature DCs to wild-type HSV-1 expressing LAT (LAT(+) virus) interfered with their phenotypic and functional maturation compared to LAT(−) virus. In vitro, DCs infected with LAT(+) viruses stimulated less HSV-specific CD8+ T cells than did DCs infected with LAT(−) virus. This could logically lead to lower HSV-specific CD8+ T-cell responses in latently-infected TG, since DCs are present in latently-infected TG. Since functional HSV-specific CD8+ T cells appear to be an important immune surveillance mechanism in decreasing reactivation from latency (1,2,15
–18,52
–54), the relatively lower HSV-specific CD8+ T-cell responses observed in TG latently-infected with LAT(
In some reports (24 –26,55), HSV-1 infection inhibits maturation of DCs similarly to the inhibition shown in this report for LAT(+) viruses. In other reports (26,32), inhibition of DC maturation was not observed. However, since the HSV-1 strains, the mutants, the MOIs, and the durations of infection differed in these studies, direct comparisons are difficult. Using the McKrae strain of HSV-1 and its corresponding mutants, our results suggest a direct or indirect suppressive mechanism on DC maturation by LAT. In addition, unlike the virion host shutoff protein (vhs), which functions to destabilize total mRNA/proteins (55), the effect of LAT was not global. Indeed, while cell surface expression of MHC, CD80, and CD86 molecules on DCs were impaired, there was no effect on the expression of other molecules, such as CD11c or CD40. This indicates that LAT-mediated impairment of cell surface expression is specific to MHC, CD80, and CD86 molecules exclusively. Our results are in agreement with reports suggesting that HSV-1 decreases DC maturation (26,30 –32,55,56). To our knowledge, the results reported here represent the first evidence that LAT may be involved directly or indirectly in this reduced DC maturation.
We found that the expression of LAT in HSV-1-infected DCs specifically prevented production of the pro-inflammatory cytokines IL-6, IL-12, and TNF-α, following stimulation with LPS. IL-12 is critically involved in the generation of cellular immunity to HSV-1 (41,42). DCs have been identified as the major producers of IL-12 in lymph nodes, thus driving T-cell immunity towards a Th-1-type response, which is critical in protection against HSV-1 infection (41,42). The inhibitory effect of HSV-1 on IL-12 production by DCs has also been recently observed in a different experimental setting (23,32). Our results confirm and extend those findings by demonstrating that LAT enhances that ability of HSV-1 to interfere with IL-12 production in DCs. If LAT also reduces the production of proinflammatory cytokines in response to HSV-1 infection in vivo, it may modulate the Th-1 cell response, and help the virus reactivate by evading TG-resident CD8+ T-cell immunosurveillance.
We report for the first time that a small number of HSV-antigen-positive DCs are present in latently-infected TG. This does not imply that TG-resident DCs were actually infected with HSV-1 or harbored latent virus. When and where the DCs acquired HSV-1 Ags, either through HSV-1 infection of DCs, through cross-presentation mechanisms by which DCs acquired Ags from necrotic or apoptotic HSV-1-infected neurons, or through other mechanisms, are important questions for future studies. Kinetic studies will be required to determine when and where TG-resident DCs become positive for HSV-1 Ag. It will also be of interest to determine if the HSV-1 Ag-positive DCs acquired virus or Ag at the periphery and then migrated to the TG, or whether these DCs were resident in the TG and acquired virus or Ag within the TG. In the latter case, it will be of interest to determine if the HSV-1 Ag-positive DCs acquired HSV-1 virus or Ag prior to or after establishment of latency. In addition, since only a very small number of HSV-antigen-positive DCs (∼6–15) appear to be present in latently-infected TG, sorting these cells for functional analysis, co-localization studies of H2b and herpes antigens, or RT-PCR was not feasible.
CD8+ T cells appear to be important in monitoring HSV-1 reactivations (13,17). During HSV-1 neuronal latency in TG of mice and humans, some neurons are surrounded by CD8+ T cells (52,57 –61). Since CD8+ T cells are presumably attracted to these neurons by viral Ags, it is assumed that the neurons surrounded by CD8+ T cells are those in which the virus has initiated the early stages of reactivation from latency. In mice, experimental reactivation of HSV-1 from latency is typically accomplished by explanting TG into tissue culture media for up to 14 days, and testing for the appearance of infectious (i.e., reactivated) virus. In this TG explant-induced reactivation model, depleting CD8+ T cells with specific mAbs leads to the detection of more reactivated virus (57). Conversely, the addition of exogenous CD8+ T cells reduces detection of reactivated virus (17,62). Thus, with wild-type HSV-1, CD8+ T cells in the TG are apparently able to reduce the detection of infectious reactivated virus.
We recently showed that during latency the function of HSV-specific CD8+ T cells is partially impaired in the TG of mice that had been ocularly infected with LAT(+) HSV-1 compared to LAT(−) HSV-1 (McKrae strain) (1,2). In mice latently infected with the KOS strain of HSV-1 via flank scarification, Mackay et al., more recently showed that HSV-specific CD8+ T cells derived from sensory dorsal root ganglia (DRG) do retain the functional ability to respond to local challenge with wild-type virus (14). However, that elegant study did not compare CD8+ T-cell function in the DRG or TG of mice latently infected with LAT(+) versus LAT(−) HSV-1. The finding in the present study that compared to LAT(−) HSV-1, LAT(+) HSV-1 interferes with DC maturation, suggests a mechanism by which LAT may reduce CD8+ T-cell function in the TG of HSV-1-infected mice. In other systems it is well known that a high-sustained Ag load can lead to CD8+ T-cell exhaustion (63 –66). However, during latency in mouse TGs, very few neurons have detectable viral Ag by immunostaining (1,2,67,68). The amount of latency and detectable viral Ag in sensory neurons of TG from mice latently infected with LAT(−) virus is even lower than with LAT(+) virus (1,2). Consistent with this, we found that there were more HSV-1 Ag-positive DCs in the TG of CD11c/eYFP transgenic mice latently infected with LAT(+) virus, compared to LAT(−) virus. This is another mechanism by which LAT may increase CD8+ T-cell exhaustion in latently infected TG. Thus, even though CD8+ T cells are much more sensitive to Ag than the antibodies used for immunostaining, the very low un-sustained Ag level that appears to be the situation in TG-resident DCs may result in the exhaustion of CD8+ T cells. Thus, it seems that the CD8+ T-cell exhaustion could be due to the viral Ag load from DCs, unless additional factors contribute to immune stimulation. For example, TG-resident HSV-specific CD8+ T cells could have a higher functional avidity (ability to respond to low epitope density) than their counterparts in the periphery (15). Alternatively, CD8+ T-cell exhaustion may suggest that there is a lot more viral Ag present in the TG of mice latently infected with LAT(+) virus than has previously been thought, or that there is a lot of undetected viral reactivation in the TG of mice latently infected with LAT(+) viruses.
The consequences of LAT interfering with DC maturation, and LAT resulting in increased DC Ag load, may both lead to decreased CD8 T-cell function. This may have implications for the immune evasion of the virus and its reactivation in latently infected TG. As illustrated in Fig. 6, if HSV-1-specific CD8 T cells in the TG help prevent initiation of viral reactivation, then LAT's ability to interfere with DC maturation may be involved in LAT's ability to increase viral reactivation. Thus, LAT's ability to cause significantly more phenotypic and functional exhaustion of CD8 T cells in latently infected TG (1,2), combined with LAT's ability to interfere with DC maturation, may constitute a significant immune-evasion mechanism, which results in increased viral reactivation from latency.
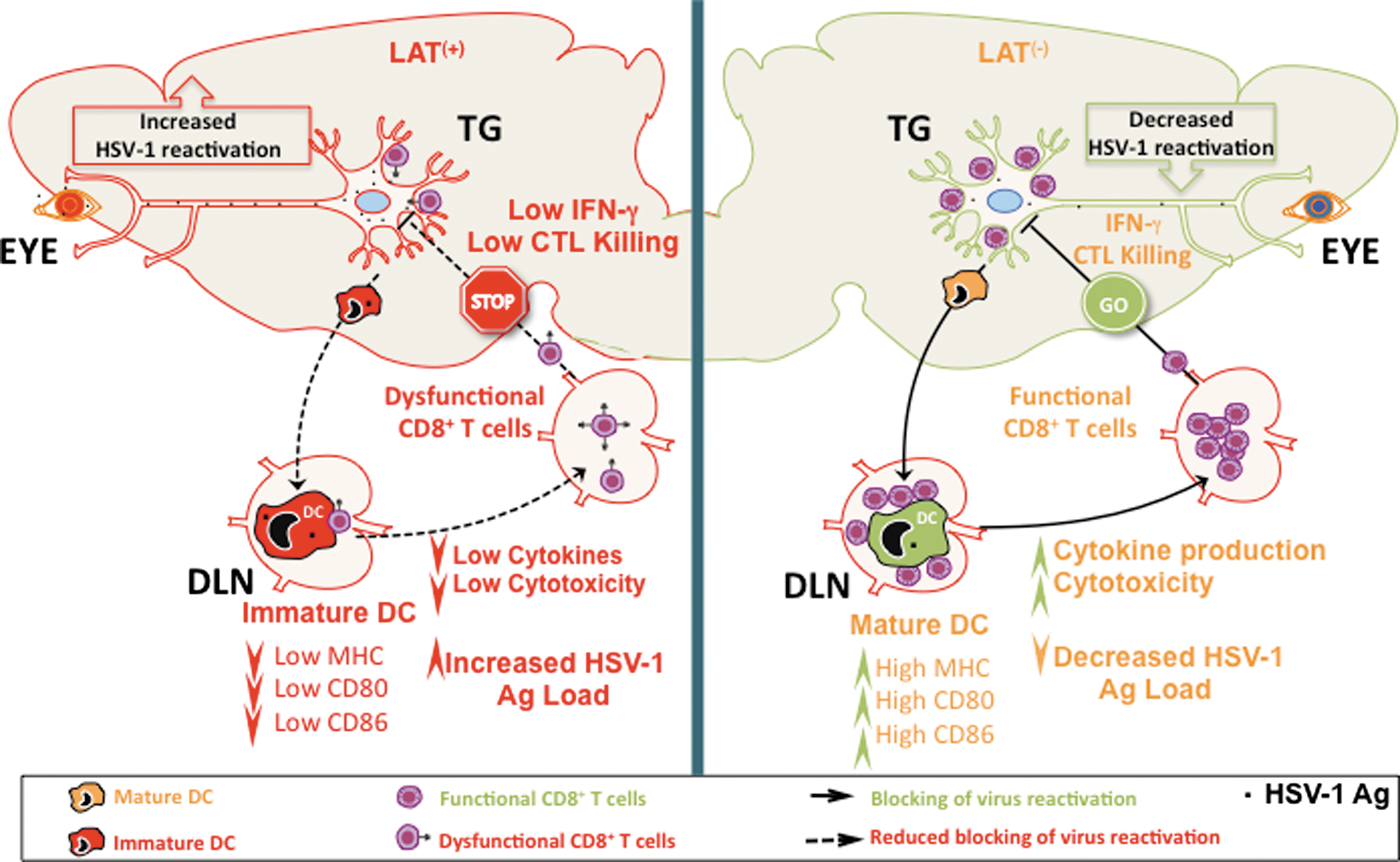
Proposed model explaining the immune evasion mechanisms whereby the HSV-1 LAT interferes with DC maturation and promotes dysfunctional HSV-specific CD8+ T cells, resulting in more virus reactivation (see text for details). Immature DCs (red) exert sentinel functions at peripheral sites, including the latently-infected TG, and are highly efficient in antigen capture, but poor in stimulating T cells. In contrast, mature DCs (peach color), are highly efficient in presenting processed antigen to naive CD8+ T cells, and promote expansion of memory CD8+ T cells. While similar numbers of DCs exist in LAT(+) and LAT(−) TG, unlike DCs infected with LAT(−) virus, DCs infected with LAT(+) viruses have less cell surface expression of MHC class I and the co-stimulatory molecules CD80 and CD86, along with impaired production of IL-6, IL-12, and TNF-α. Consequently, LAT(+) immature DCs, but not LAT(−) DCs, are unable to stimulate HSV-specific CD8+ T-cell function. Thus CD8+ T cells in the LAT(+) TG have less cytotoxic activity (CTL killing), and cytokine (IFN-γ) production, which would decrease virus reactivation from sensory neurons. These results suggest a novel immune-evasion mechanism whereby HSV-1 LAT interferes with DC maturation to indirectly escape immunosurveillance by TG-resident CD8+ T cells, and consequently increases viral reactivation. Color images available online at
Although LAT does not appear to make any abundant protein, it does encode 2 small RNAs with anti-apoptosis activity (69), and 8 miRNAs (70,71). One or more of these RNAs may be involved in how LAT interferes with DC maturation and CD8 T-cell exhaustion. However, investigation of these possibilities is beyond the scope of the present study.
In summary, we have shown here, for the first time, that LAT interferes with phenotypic and functional maturation of DCs. Either or both of these LAT functions may represent a LAT-mediated immune-evasion mechanism, whereby LAT indirectly leads to HSV-specific CD8+ T-cell exhaustion in latently infected TG. This would represent a LAT function, in addition to its ability to block apoptosis (19), that may play an important role in increasing reactivation of HSV-1.
Footnotes
Acknowledgments
This work was supported by Public Health Service NIH grants EY14017, EY14900, and EY019896 to L.B.M., EY013191 and EY018171 to S.L.W., The Discovery Eye Foundation, and a Research to Prevent Blindness Challenge grant. L.B.M. is an RPB Award Investigator. The authors would like to thank Dr. Douglas McAlester (ViroStat) for kindly sharing herpes monoclonal antibodies, and Dr. James V. Jester and Dr. Donald Brown for assistance with the confocal microscopy studies. We also would like to thank Dr. Alison Deckhut Augustine and Amy K. Stout from the NIH Tetramer Facility for providing the tetramers used in this study.
Author Disclosure Statement
No competing financial interests exist.