
Abstract
We have evaluated the role of γ-secretase, which is a crucial component in the Notch-induced signaling cascade, on herpes simplex virus type 2 (HSV-2)-induced innate and acquired interferon responses in human CD4+ T cells and plasmacytoid dendritic cells (pDC). We found that blockade of the Notch signaling pathway with a pharmacological γ-secretase inhibitor blocked both HSV-2-induced interferon-γ (IFN-γ) production in CD4+ T cells, and HSV-2-induced IFN-α production in pDC in a dose-dependent fashion. These effects were not due to an overall suppressive capacity of the γ-secretase inhibitor, as it affected neither phytohemagglutinin (PHA)-induced IFN-γ production in CD4+ T cells, nor CpG-induced IFN-α production in pDC. Our data suggest that Notch signaling could be involved in HSV-2-induced interferon responses in CD4+ T-cells and pDC.
Introduction
Plasmacytoid dendritic cells (pDC) are the main type I IFN producers in the body (14), and they produce high levels of IFN-α in response to HSV-2 by the binding of genomic DNA from HSV-2 to TLR9 (13,14). Fibroblasts and macrophages also express type 1 IFN, and in these cells the recognition of viral infection is mediated by the retinoic acid-inducible gene-I (RIG)-I together with TLR9, a process that is also dependent on RNaseL (24,25). Other factors known to induce IFN production after HSV binding include the mannose receptor (15,26), and the chemokine receptors CCR3 and CXCR4 (1). Type II IFN, on the other hand, is mainly secreted by natural killer cells and T cells (3,27).
Notch proteins (Notch1−4) are transcriptional activators expressed in a wide range of immune cells (23). Following interaction with either of its ligands, the Notch/ligand complex is cleaved first by a metalloprotease, and then by γ-secretase. These processes generate an active intracellular portion of Notch (NotchIC), that can translocate to the nucleus and act as a transcription activator (7,8,17). Notch proteins are involved in the development of a wide range of fetal and postnatal processes, and their role in hematopoiesis and thymocyte development are well documented (23). More recent studies also address a role for the Notch proteins in the regulation of peripheral T-cell responses, including CD4+ T-cell activation and IFN-γ induction (20). In this study we wanted to investigate whether Notch signaling is involved in HSV-2-specific IFN responses in CD4+ T cells and in pDC.
It has previously been shown that Notch signaling can be blocked with a γ-secretase inhibitor, and that this will decrease the IFN-γ production in CD3/CD28-stimulated CD4+ T cells under neutral or Th-1-driving conditions (16). We verified these results and showed that the CD4+ T-cell response to HSV-2 can be blocked by inhibiting γ-secretase cleavage in the Notch-signaling pathway.
First, we confirmed the expression of Notch1 on PBMC. As expected, the Notch1 receptor was expressed on the surface of CD4+ T cells (Fig. 1A). To assess the role of γ-secretase signaling in HSV-2-mediated IFN-γ responses, purified CD4+ T cells were stimulated with irradiated HSV-2 in the presence or absence of the γ-secretase inhibitor compound E (CalBiochem, San Diego, CA) at different concentrations. The γ-secretase inhibitor completely blocked the production of IFN-γ from CD4+ T cells, while the non-treated cells produced IFN-γ, albeit at different levels, in response to HSV-2 (Fig. 1B). This inhibition was dose-dependent, since CD4+ T cells stimulated with the γ-secretase inhibitor were totally incapable of producing any IFN-γ at inhibitor concentrations higher than 5.6 μM (range 50–5.6 μM; Fig. 1C). At lower concentrations (1.8 μM), the amount of IFN-γ produced was comparable to that seen in cultures without any inhibitor added (Fig. 1C). The inhibition of IFN-γ production in HSV-2-activated CD4+ T cells was not due to an overall suppressive ability of the γ-secretase inhibitor, as seen after CD3/CD28 stimulation (16), since PHA-induced IFN-γ production in CD4+ T cells remained unaffected after culture with the inhibitor (Fig. 1D). This indicates that HSV-2-mediated IFN-γ production is dependent on the Notch-signaling pathway, suggesting a new, previously unknown intracellular signaling pathway downstream of the Notch cascade in the CD4+ T-cell response against HSV-2 infection. Blockade of γ-secretase cleavage under Th-1-polarizing conditions inhibits the activation of the transcription factor T-bet, which leads to diminished IFN-γ production (16). It is possible that the abolished IFN-γ production seen after γ-secretase blockade of HSV-2-activated CD4+ T cells is due to the inhibition of T-bet in a similar manner, since T-bet is essential to HSV-2-induced immunity (30). However, this remains to be evaluated.
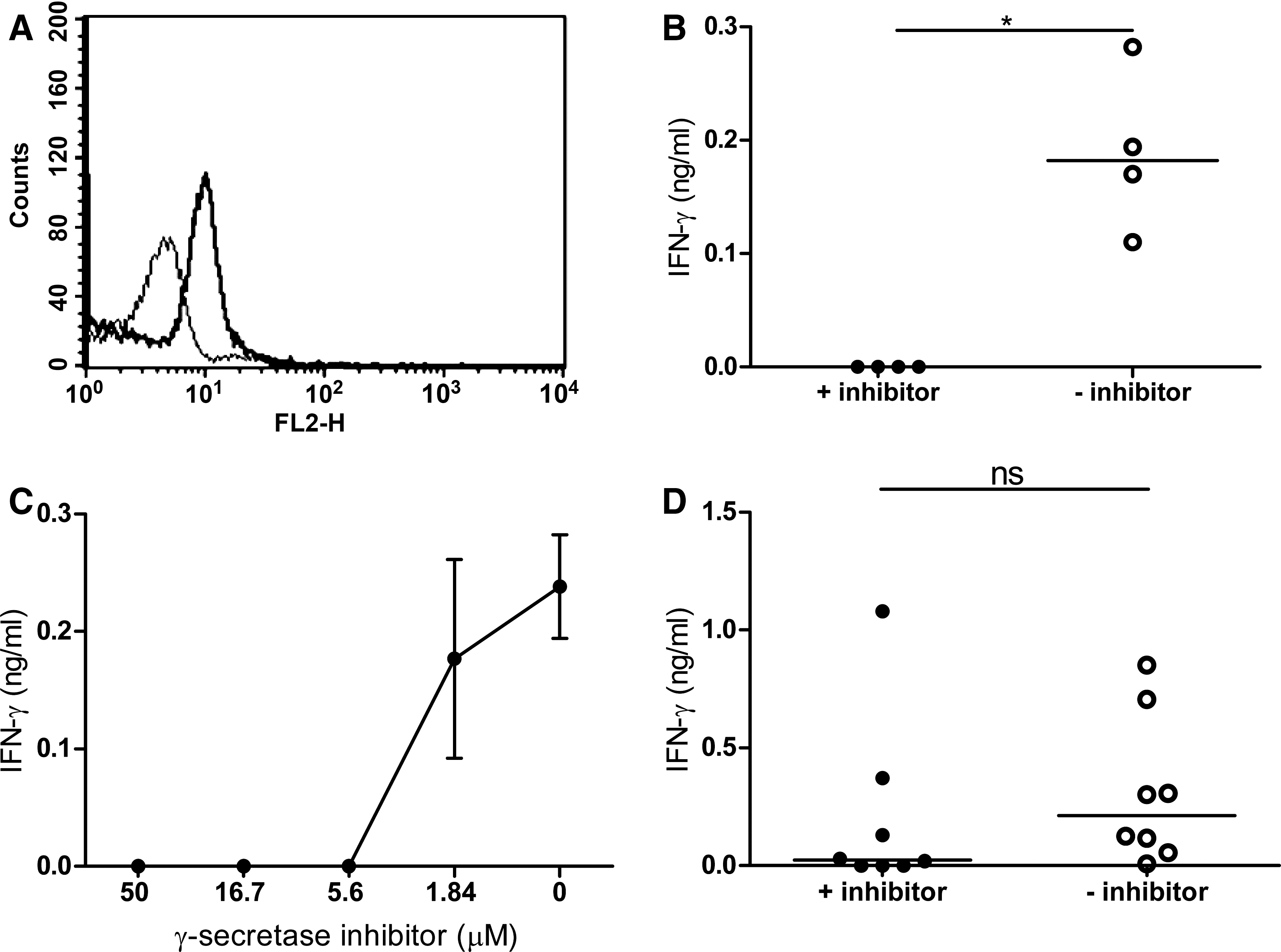
Inhibition of γ-secretase cleavage blocks HSV-2-induced IFN-γ production in CD4+ T cells.
Dontje et al. have previously shown that Notch1 is expressed on CD34+ precursor cells, and that the level of Notch1 expression decreases when the precursor cells differentiate into pDC (4). Interaction of Notch1 on CD34+ progenitor cells with its ligand delta-1 leads to the maturation of pDC, a development that can be blocked with a γ-secretase inhibitor (19). Here, we show that mature pDC also express the Notch1 receptor (Fig. 2A), and that their IFN-α production is almost completely abolished after blockade of the γ-secretase cleavage step in response to HSV-2 (Fig. 2B). It has previously been shown that HSV-2 can induce IFN-α production in murine pDC, by the binding of genomic viral DNA to TLR9, and signaling through MyD88 (13). We confirm that pDC produce IFN-α in response to irradiated HSV-2 (Fig. 2B), via TLR9 ligation (Fig. 3A). TLR9 signaling is induced by the binding of CpG-rich motifs to the receptor, which gives rise to high levels of IFN-α. The stimulatory effect of CpG can be blocked with a G-ODN that is characterized by the presence of a repetitive sequence of five guanosines and no stimulatory CpG motifs (22). Indeed, the stimulatory effect of CpG in our system was blocked after stimulation with CpG and G-ODN (Fig. 3B). More importantly, we also show that HSV-2 strain 333 signals through TLR9, since pDC stimulated with the virus together with G-ODN produced reduced amounts of IFN-α (Fig. 3A). However, the IFN-α production from HSV-2-stimulated pDC was almost completely abolished by the γ-secretase inhibitor (Fig. 2B). Similarly to the inhibition of IFN-γ secretion in CD4+ T cells, the inhibition of IFN-α production also occurred in a dose-dependent fashion (Fig. 2C). pDC cultured with high doses of the γ-secretase inhibitor (100 μM) produced very low amounts of IFN-α, which increased with decreasing concentrations (100–25 μM) of the inhibitor (Fig. 2C). The inhibition of IFN-α production was particularly impressive in HSV-2-exposed pDC (Fig. 2B), whereas the CpG-induced IFN-α production was less affected, perhaps due to the lower IFN-α responses obtained with CpG (Fig. 2D). This indicates that the Notch/γ-secretase pathway might be involved in the HSV-2-induced IFN-α production. Thus, both the Notch/γ-secretase pathway and the TLR9/IRF-7/MyD88 pathways (11,12) might be required for adequate HSV-2-induced IFN-α responses in pDC. This supports the notion that the type I IFN pathways are complex, and involve several pathways that interact both prior to, and during, the IFN response.
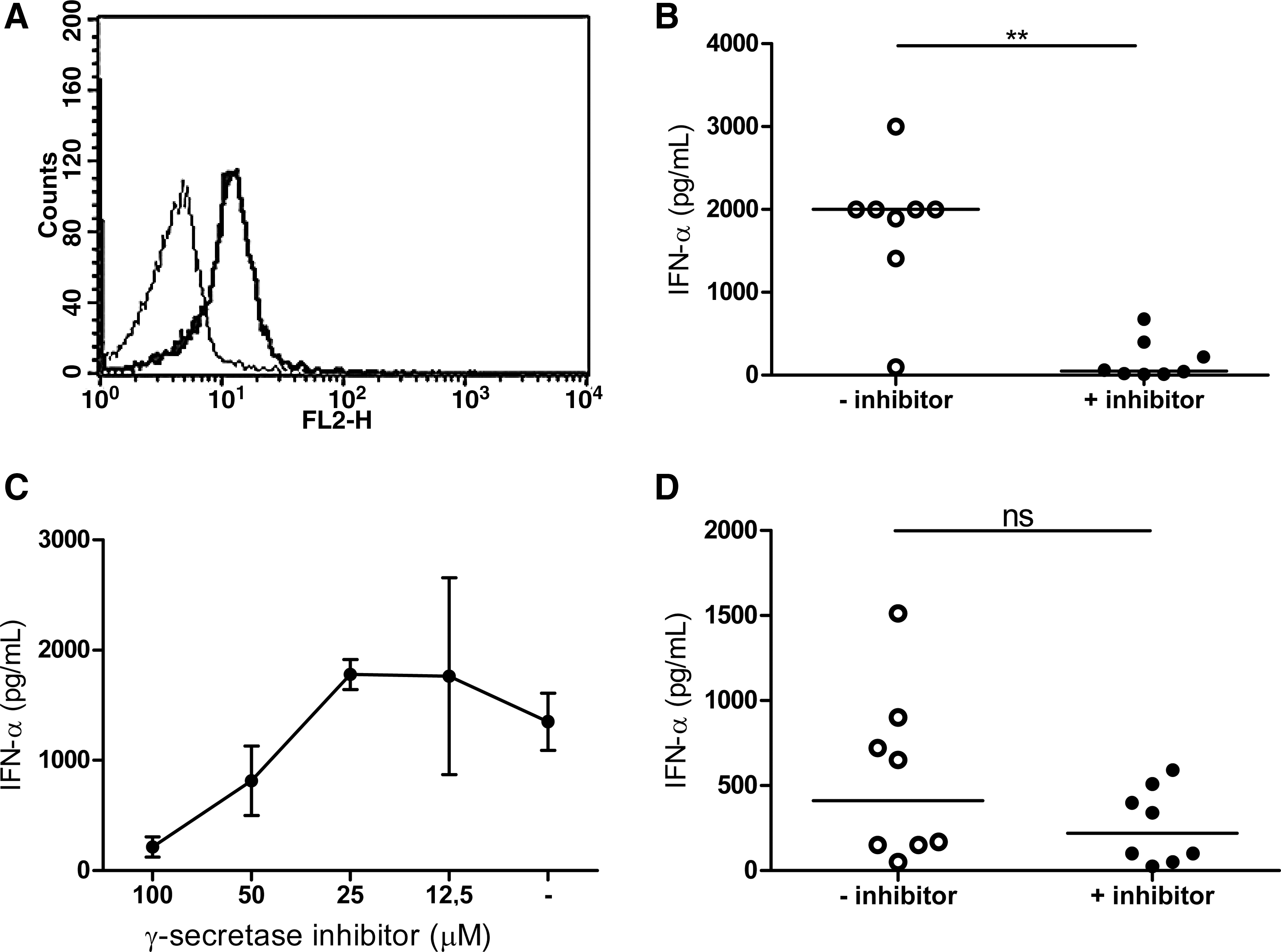
Inhibition of γ-secretase cleavage blocks HSV-2-induced IFN-α production in pDC.
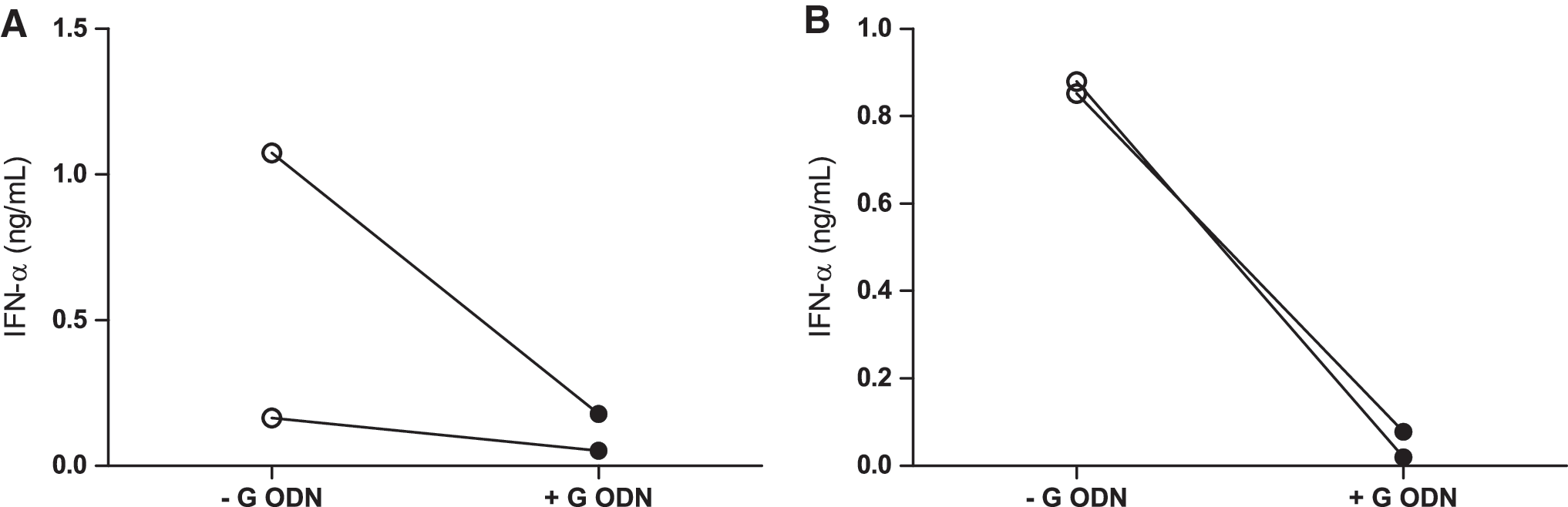
Blockade of TLR9 inhibits HSV-2-
In conclusion, we show that blocking Notch signaling through the administration of a γ-secretase inhibitor completely blocks IFN-γ, and clearly diminishes IFN-α production in CD4+ T cells and pDC, respectively. Both type I and type II IFNs are essential to combat HSV-2 infection (5,18,28,29), and blockade of the γ-secretase signaling pathway may increase both the risk and the severity of genital HSV-2 infection. This is important to take into consideration in the medical therapy of, for example, Alzheimer's patients, in whom γ-secretase inhibitors are commonly used as treatment. Thus we suggest that Notch signaling, via the cleavage of γ-secretase, could be involved in HSV-2-induced interferon responses in both CD4+ T cells and in pDC, via yet unknown downstream pathways.
Footnotes
Author Disclosure Statement
No competing financial interests exist.