
Abstract
Background:
Soft ticks (Family: Argasidae) are vectors of relapsing fever Borrelia in the United States and are potential vectors of African swine fever virus, a pathogen that could have a devastating effect on the U.S. swine industry if introduced to the U.S. mainland. Much of the tick-borne disease research in the U.S. focuses on hard ticks, and less is known about the ecology of soft ticks. Some soft tick species found in the southern U.S. have a wide host range and may feed on cattle, swine, native and exotic ungulates, small mammals, reptiles, and humans. Because the feeding habit of most soft tick species involves taking short, repeated blood meals that may include multiple host species, pathogen transmission among hosts is a concern both for human and animal health.
Materials and Methods:
Sampling was carried out at four locations in south Texas using dry ice traps placed in or near animal burrows and other sheltering cracks and crevasses that may provide refuge for soft ticks. Collected ticks were identified and subsequently screened for Rickettsia and Borrelia species and for host bloodmeal detection using conventional polymerase chain reaction and Sanger sequencing for pathogen and host species identification.
Results:
In total, 256 ticks of two Ornithodorinae species were screened. Borrelia species were identified in three samples. Bloodmeal detections were made in 22 tick specimens, representing eight vertebrate host species.
Conclusions:
Results demonstrate that the soft tick species detected herein feed on a range of wildlife hosts in south Texas and are associated with agents of human disease.
Introduction
Soft ticks (Ixodida: Argasidae) feed on a wide range of vertebrate species and are vectors of pathogens affecting humans and other animals (Butler and Gibbs 1984; Dworkin et al., 2002; Donaldson et al., 2016; Piccione et al., 2016; Guzmán-Cornejo et al., 2019). In the United States, many soft tick species are associated with wildlife hosts, but may also feed on humans, livestock, and other domestic animals (Cooley and Kohls 1944). Previously unidentified rickettsial species or rickettsial species of unknown pathogenicity have been identified in soft ticks, including R. hoogstraali in soft ticks in the United States (Latas et al., 2020) and a novel Rickettsia species detected in Ornithodoros erraticus in Portugal (Milhano et al., 2014). Ornithodoros species are associated with the transmission of relapsing fever agents to humans (Bates et al., 1921; Cooley and Kohls 1944, Dworkin et al., 2002). Particularly in the state of Texas, transmission of Borrelia turicatae, one agent of human relapsing fever, is associated with encounters with soft ticks in caves and in primitive lodgings (Bissett et al., 2018; Busselman et al., 2021; Christensen et al., 2017; Dworkin et al., 2008).
Soft ticks remain relatively understudied compared with their hard tick counterparts. Because many soft tick species take numerous, rapid bloodmeals, they are rarely collected on host animals (Donaldson et al., 2016). Little is known about the biology and ecology of many native tick species in south Texas, including soft ticks such as Ornithodoros species. In Texas, exotic mammals are commonly stocked for the purpose of sport hunting (Lohmeyer et al., 2018), and interactions between native tick species and exotic host species may pose a pathogen transmission concern. For example, African warthogs are a reservoir host for African swine fever virus (ASFV), which causes devastating disease in domestic swine (Golnar et al., 2019; Wormington et al., 2019). In endemic areas, Ornithodoros spp. are associated with maintenance and transmission of ASFV (Butler and Gibbs 1984), and Ornithodoros species native to the United States have demonstrated vector potential in laboratory experiments (Hess et al., 1987). Although ASFV has not been introduced into the United States, introduction and the subsequent spread to native ticks, domestic swine, and feral swine is a biosafety concern (Butler and Gibbs 1984; Golnar et al., 2019; Wormington et al., 2019). The escape of African warthogs from high-fence ranches has resulted in the establishment of feral populations in areas of south Texas (Mayer et al., 2020). It is unclear if or how soft ticks may be interacting with exotic wildlife species.
Bloodmeal analysis has been used to examine the host use by blood-feeding arthropods including mosquitoes, soft ticks, and triatomines (Cupp et al., 2004; Busselman et al., 2021; Balasubramanian et al., 2022; Reeves et al., 2018). Bloodmeal analysis relies on the detection of residual host DNA after arthropod feeding. Because many soft tick species feed multiple times within nymphal and adult life stages, bloodmeal analysis can be used to examine host use (Apanaskevich and Oliver 2014; Busselman et al., 2021). The purpose of this research was to examine pathogen prevalence in soft tick species collected in south Texas and to evaluate soft tick host use through bloodmeal analysis.
Methods
Sampling for soft ticks was conducted from June through November 2022 at three locations in south Texas (Fig. 1). The Chaparral Wildlife Management Area (WMA) in Dimmit and La Salle counties, where feral populations of African warthogs are known to occur, was sampled from June through October at weekly or biweekly intervals for a total of 11 trapping sessions. During the first two trapping sessions daytime sampling was conducted, while overnight sampling occurred during the remaining sessions due to extreme daytime temperatures. Lake Casa Blanca International State Park near Laredo, TX in Webb County was sampled overnight twice; once in the 1st week of October; and once in the 3rd week of October. Overnight sampling was conducted at the McAllen Nature Center in McAllen, TX in Hidalgo County 31 October–01 November.
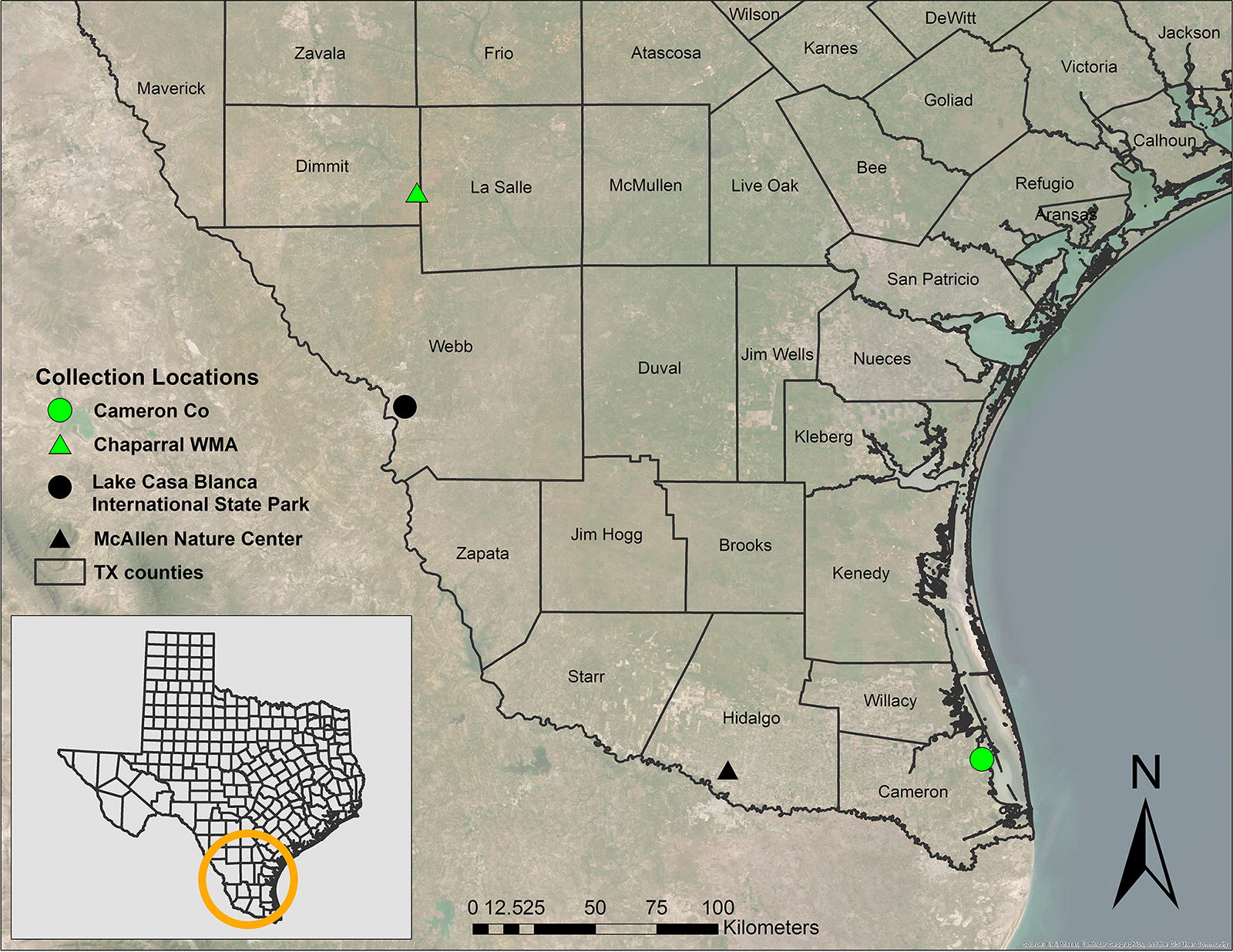
Sites of tick collection in southern Texas. Green icons indicate locations at which ticks were collected. Black icons indicate locations at which ticks were not collected. Inset map of the state of Texas with circle indicating the south Texas sampling area. Service Layer Credits: Source: Esri, Maxar, Earthstar Geographics, and the GIS User Community.
Sampling of animal burrows and sheltered cracks and crevasses likely to provide refuge to soft ticks was accomplished using modified dry ice traps, each consisting of a 1.9L cooler filled with dry ice, which sublimated to produce carbon dioxide to attract ticks. One end of a 1.9 cm diameter polypropylene tube approximately 0.6 m in length was attached to the mouth of the cooler, and the other was affixed to a cylindrical sticky trap approximately 7.5 cm in length. Masking tape was affixed to the surface of the sticky trap with the sticky side facing outward, from which adhered ticks could be removed without requiring the removal of residual adhesive. The sticky traps were placed into the openings of animal burrows, hollows beneath tree stumps and rock formations, and beneath wooden and rock pavilions and structures at the various sampling locations. Upon trap retrieval, captured ticks from each trap were removed from the tape with forceps and placed in 95% ethanol. Samples were placed in a cooler with dry ice during transport from collection sites and stored until extraction at −20°C.
Collected ticks were morphologically identified to life stage and species (Cooley and Kohls, 1944; Guzmán-Cornejo et al., 2019; Pratt and Stojanovich, 1963). The classification scheme of Mans et al. (2021) and Kneubehl et al. (2022) was used, in which members of Carios are reclassified into their former subgenera, and Alectorobius is elevated to genus.
Prior to DNA extraction, ticks were surface sterilized using a serial dip in 10% bleach, UltraPure H2O, and 80% EtOH, and then ethanol was allowed to evaporate for ∼20 min. Each tick was laterally bisected with a sterilized scalpel blade and subjected to a three-hour Proteinase K digest. Whole genomic DNA was extracted using a Mag-Bind® Blood and Tissue kit (Omega Bio-tek, Inc., Norcross, GA) following the manufacturer-provided protocol.
Screening for relapsing fever Borrelia and spotted fever group Rickettsia species was conducted using polymerase chain reaction (PCR) with primers targeting the IGS and GlpQ genes (Latas et al., 2020) for Borrelia spp. and the OmpA gene for Rickettsia spp. (Latas et al., 2020; Raoult et al., 2001). The IGS primers were used for initial screening, and the GlpQ primers were used for confirmation and sequencing of Borrelia-positive samples (Latas et al., 2020). Borrelia lonestari and Rickettsia amblyommatis DNA were used as positive controls for pathogen screening. Confirmatory sequencing of a select Borrelia-positive sample was carried out using primers targeting the 16S, IGS, and flaB genes (Bermúdez et al., 2021; Marti Ras et al., 1996). Primers targeting the 12S and COI gene of the tick were used for the same sample to confirm tick species identity (Lv et al., 2014). The resulting Borrelia sequences were deposited in GenBank under accession numbers PP812536-PP812538, PP824842, PP827027, and PP831614.
Bloodmeal analysis was conducted using three primer sets, with one targeting the COI gene (Reeves et al., 2018), and two targeting the CytB gene: “mammal c” (Busselman et al., 2021) and “Herp1/Herp2” (Balasubramanian et al., 2022). Although some of the primer sets were initially developed to target specific taxa (e.g., the “Herp1/Herp2” primer set), they have been reported to amplify a much broader range of vertebrate species (Balasubramanian et al., 2022; Busselman et al., 2021). DNA extracted from bovine blood was used as a positive control for blood-meal identification.
PCR amplicons were visualized using gel electrophoresis. Positive samples were sent for bidirectional Sanger sequencing by Eurofins Genomics, LLC (Louisville, KY). Sequences were assembled and cleaned using GeneStudio™ Pro v2.2 (GeneStudio, Inc.) and compared with existing sequences in the NCBI database using the Basic Local Alignment Search Tool (BLAST) to identify the host bloodmeal. In some instances, particularly with the “Herp1/Herp2” primer set, both forward and reverse sequence reads were successfully produced, but could not be assembled; if resequencing attempts did not resolve the issue, the forward read was used for BLAST comparison.
DNA aliquots from an additional 143 Alectorobius talaje (Guérin-Méneville) were obtained from USDA-ARS Cattle Fever Tick Research Unit (CFTRU) personnel for pathogen and bloodmeal screening. These ticks were collected by CFTRU personnel from a total of 14 burrows at two locations in Cameron County, TX between February 2022 and March 2023 on conventional dry ice traps placed near animal burrows. Collected ticks were stored in 90% ethanol at room temperature prior to DNA extraction. Following surface sterilization, ticks were homogenized using an Omni Bead Ruptor 24 (Omni International, Inc., Kennesaw, GA). Whole genomic DNA was extracted with a DNeasy® Blood and Tissue kit (Qiagen, Valencia, CA) following a 3-hour Proteinase K digest. PCR was conducted as described above.
Results
In total, 79 nymphs and 34 adults were collected from the Chaparral WMA, from 26 of 130 trap locations between June 28 and October 25, 2022. Seventy-four ticks (53 nymphs and 21 adults) were identified as Ornithodoros turicata (Dugès), and 39 ticks (26 nymphs and 13 adults) were presumptively identified morphologically as Alectorobious talaje (formerly Ornithodoros [Alectorobius] talaje) based on current available information for adults of the species. A subset of 22 live-collected ticks from the Chaparral WMA sent to the Texas A&M Tick Research Laboratory for colony establishment was identified by Tick Research Laboratory personnel as O. turicata and A. talaje. No ticks were collected from Lake Casa Blanca International State Park or the McAllen Nature Center.
Three ticks collected from the Chaparral WMA, two O. turicata nymphs and one A. talaje nymph, were infected with Borrelia species identified with the glpQ primer set. The two O. turicata specimens were infected with B. turicatae that was 100% identical to B. turicatae isolated from a domestic dog in Florida, USA (GenBank AY934640). In one of the two samples, only the reverse sequence was successfully amplified during sequencing, but yielded a clean sequence with well-defined peaks. A second attempt at sequencing in the forward direction was unsuccessful. The A. talaje specimen was infected with a Borrelia species that was 99% similar to B. puertoricensis isolated from A. puertoricensis in Panama (GenBank MZ229856) when sequenced with the glpQ primer set. The next closest identified species match was Borrelia parkeri, with 98% identity (GenBank MH704900). The sample shared 97% identity with B. turicatae (GenBank CP129306) and Borrelia venezuelensis (GenBank MG651651).
Additional amplification and sequencing with primers targeting flaB, IGS, and 16S gene regions yielded results that were 96–99% identical to B. puertoricensis (Table 1). The amplicon generated with the flaB primer set was 100% identical to a species (Candidatus Borrelia texasensis) that was identified from a Dermacentor variabilis specimen collected in south Texas and thought to represent a previously undescribed relapsing fever Borrelia (Lin et al., 2005). No ticks from the Chaparral WMA were infected with Rickettsia species.
Relevant Sequence Matches for Amplicons Generated with Multiple Primer Sets for Identification of Borrelia Spp. Detected in Soft Ticks in South Texas and for Molecular Identification of a Tick Morphologically Identified as Alectorobius talaje
Where a BLAST search revealed multiple high similarities to the amplified gene regions for the Borrelia species detected in the A. talaje specimen, information for more than one matching species is shown.
BLAST, Basic Local Alignment Search Tool.
Sequencing with primers targeting 12S and COI gene regions to verify the identity of the morphologically identified A. talaje specimen infected with Borrelia yielded few close matches, with all nearest matches belonging in the Ornithodorinae subfamily. The amplicon generated with 12S primers yielded a 99% match to a Carios sp. collected in south Texas (GenBank OK394016). The next nearest matches were various Alectorobius species at ∼84–86% identity (Table 1). The amplicon generated with COI primers was 83% identical to Alectorobius faccinii (GenBank KY678235) and 82% identical to Alectorobius puertoricensis (GenBank ON800873).
Host DNA detections with BLAST sequence identification matches of >90% with at least one primer set were made in 21 ticks from the Chaparral WMA: three O. turicata adults, 17 O. turicata nymphs, and one A. talaje nymph (Table 2). Seven vertebrate host species were identified from these ticks, although the number of host species detections for each individual primer set were variable; namely: host detections were made with all three primer sets in only one sample, detections were made with two of three primer sets in five samples, and detections were made with only one of three primer sets in 15 samples. Specific primer sets for each bloodmeal identification, the percent match to reference samples in GenBank, the sequence length, the percent query cover, and the GenBank reference sequence accession number are displayed in Table 2.
Host Blood-Meal Identification from Ornithodoros Species Collected from the Chaparral Wildlife Management Area in 2022 Using Three Vertebrate-Targeting Primer Sets
One O. turicata adult and two O. turicata nymphs contained nine-banded armadillo (Dasypus novemcinctus) DNA. Four O. turicata nymphs contained raccoon (Procyon lotor) DNA. One O. turicata adult and nine nymphs contained Texas tortoise (Gopherus berlandieri) DNA. One O. turicata nymph contained coachwhip (Masticophis flagellum) DNA. One O. turicata adult contained western diamondback rattlesnake (Crotalus atrox) DNA. One O. turicata nymph contained Texas toad (Anaxyrus speciosus) DNA. One A. talaje nymph contained wild turkey (Meleagris gallopavo) DNA. Only the reverse sequence for this sample was successfully amplified, and a second attempt to generate a forward sequence was also unsuccessful. This amplicon band was very faint on the gel and likely represents an old and highly degraded bloodmeal.
Host detection was made in one additional sample with two primer sets, although the BLAST sequence similarities were <90%. Sequence results from the COI primer set for one O. turicata nymph resulted in an 84% match to a Western skink (Plestiodon skiltonianus) (GenBank KU986179); sequence results from the CtyB “mammal c” primer set for the same sample resulted in an 88% match to a common five-lined skink (P. fasciatus) (GenBank DQ241645).
During amplification of six of the samples containing host bloodmeals with the CytB “Herp1/Herp2” primers, tick DNA was amplified and incorporated with host DNA, resulting in sequences containing both host and tick DNA that were larger than the expected 228 base pairs. In these instances, the Herp1 sequence reads were trimmed to remove tick DNA and submitted in the BLAST search. The resulting fragments of host DNA from these samples were ∼75 base pairs. This was the case for five of the seven samples in which Texas tortoise DNA was identified, and the sample in which rattlesnake DNA was identified. Subsequent reamplification and resequencing attempts yielded the same results.
None of the A. talaje acquired from Cameron County were infected with Rickettsia species. Initial Borrelia screening with the IGS primer set yielded five samples with amplicons of the expected size. However, these samples were not amplified during subsequent screening with the glpQ primer set. An attempt to sequence one of the IGS amplicons yielded a sequence for which no match could be made in GenBank. No host detections were made in these ticks with any of the three primer sets. Initial PCR amplification during bloodmeal analysis yielded amplicons in ∼45% of samples with the COI primer set and ∼30% of samples with the CytB primer set; however, sequencing of a subset (∼25% of potential positive samples) identified only arthropod DNA or sequences for which no match could be made. Approximately 20% of samples yielded amplicons with the CytB mammal c primer set. These also yielded sequences in which only arthropod DNA or human DNA was amplified, or for which no positive match could be made. Positive control amplicons of the expected size were obtained in all PCR assays for pathogen screening and bloodmeal detection.
Discussion
The prevalence of Borrelia species in O. turicata and A. talaje collected herein was lower than expected. However, the Borrelia species detected in O. turicata, B. turicatae, is associated with human disease in Texas (Bissett et al., 2018; Christensen et al., 2017). The Borrelia amplified from A. talaje shared 99% similarity to Borrelia detected in the closely related A. puertoricensis and 100% identity to a presumed novel Borrelia species detected in D. variabilis. The detection of a Borrelia species sharing high genetic similarity with other soft tick-associated Borrelia in both A. talaje and a hard tick is surprising. However, the D. variabilis in which this Borrelia was previously reported was collected from a coyote (Lin et al., 2005). Thus, it is uncertain whether detection may have resulted from ingestion of an infected blood meal from the coyote host, or disseminated infection in the tick. In Mexico, A. talaje has been associated with the transmission of a relapsing fever Borrelia to humans, B. mazzottii (Davis, 1956); however, no B. mazzottii isolate or DNA sequence data is available for comparison with this sample. The Borrelia sp. detected herein may represent detection of a Borrelia species associated with A. talaje, such as B. mazzottii, that is not currently otherwise represented in the GenBank repository, or a novel relapsing fever group Borrelia species circulating in ticks in the region.
It should be noted that the limits of detection for the primer sets used for Rickettsia and Borrelia screening are not defined. The presence of tick DNA, host DNA from bloodmeals, and other components of the tick microbiome obtained when extracting DNA from an entire tick rather than targeted tissues such as salivary glands can complicate pathogen detection and increase the potential for false negatives. Additional sampling and screening would be beneficial in identifying the prevalence of relapsing fever group Borrelia species in south Texas.
The phylogeny of soft ticks has undergone many recent revisions, and the increasing availability of genetic data has been vital to elucidating the relationships between Argasid species (Kneubehl et al., 2022). There is currently no sequence data available for A. talaje and many closely-related species, and the resulting COI sequence generated for the Borrelia-infected tick herein was only an 83% match for A. puertoricensis, a member of the A. talaje group. The 12S sequence generated for this tick was a 99% match to an individual identified as Carios sp. collected from a white-footed mouse in south Texas (Galán et al., 2022). One of the next closest matches was Alectorobius capensis, at 85% identity. The Carios sp. match was an 84% match to the same A. capensis sequence match. It is likely that the Carios sp. match from south Texas represents an individual belonging to a species that was, at the time, included within a subgenera of Carios that has since been elevated to genus.
The identity of A. talaje in the southern U.S. is of great interest, particularly given the Borrelia detection described in the tick in this study. Difficulties in distinguishing between members of various species within the A. talaje species group are well established, particularly in areas where multiple members of the complex co-occur (Bermúdez et al., 2021). Research over the last few decades has resulted in the identification of several new species once thought to represent A. talaje in Central and South America (Bermudez et al., 2021; Venzal et al., 2008). Furthermore, some of these species can only be distinguished in the larval stage (Guzmán-Cornejo et al., 2019). Species such as A. kohlsi and A. dugesi are separated from A. talaje primarily by the size of the body, mammillae, and dorsal disks and have been suggested to be synonyms of A. talaje, although there is disagreement with this assessment (Guzmán-Cornejo et al., 2019). A previous study identified morphological differences between larval specimens identified as A. talaje from Guatemala, Mexico, and the southern U.S., but the absence of any genetic data and small larval samples sizes precluded new species descriptions (Venzal et al., 2008). In light of this information, it appears likely that southern U.S. collections of A. talaje in fact may represent one or more sister species. However, without close morphological examination of larval specimens, none of which were collected in this study, and additional genetic data, there is little alternative but to continue to refer to these individuals as A. talaje. Further investigations into the identity of the detected Borrelia species and of ticks morphologically identified as A. talaje from south Texas, including efforts to produce or acquire larvae from multiple locations, are being undertaken and will be presented elsewhere. Hosts of eight vertebrate species were detected using bloodmeal analysis, including mammal, reptile, amphibian, and avian hosts. All detections except one were made in O. turicata. Texas tortoise and raccoons were the most frequently identified hosts of O. turicata, although all individuals containing raccoon DNA and all but one individual containing Texas tortoise DNA were collected from the same burrows, indicating that these individuals may have fed upon the same host individual. One tick containing Texas tortoise DNA was collected from the same burrow from which the three ticks containing nine-banded armadillo DNA were collected. This was the only instance where ticks containing bloodmeals from two different host animals were collected from the same burrow.
Host DNA was detected in only one A. talaje specimen (n = 182). The collection method, extraction method, and storage prior to extraction of A. talaje obtained from Cameron County differed from the methods used for ticks collected from the Chaparral WMA. DNA quantification in a subset of samples revealed an average yield of 13 ng/uL for nymph and adult samples combined. Subsequent efforts to amplify tick DNA in selected samples have been successful. Detection in a small proportion of the Chaparral WMA A. talaje (1/39), which were handled in the same way as the Chaparral WMA O. turicata specimens in which host amplification was higher, suggests a cause for the lack of host bloodmeal detections unrelated to the sample processing procedures and perhaps related to the ecology of the species. Reports of this species in the United States describe it being associated with rodent hosts or human dwellings infested with rodents (Cooley and Kohls, 1944; Davis, 1940). The lack of host detections in A. talaje may indicates a lower abundance of preferred hosts and fewer incidences of feeding than in the O. turicata collected from the same site. The limits of detection for host DNA with these primer sets are unknown, and soft ticks have been reported to survive for periods of up to several years in the absence of bloodmeals (Donaldson et al., 2016). The time between intermittent bloodmeals of these ticks and subsequent digestion/degradation of bloodmeals may exceed the limits of detectability of these primer sets. It is also unknown if recently blood-fed individuals are attracted to the CO2-baited traps. This is notable, as the collection method for ticks in Cameron County involved the use of conventional dry ice traps placed near burrows, requiring ticks to leave the burrows in response to the CO2 stimulus. The modified CO2-baited traps used in other locations were placed directly in animal burrows for tick collection.
With the exception of the Plestiodon species, the identified hosts are distributed throughout south Texas (Dixon, 2013; Sibley, 2003; Schmidly, 2004). The low percentage match and similarity with two different Plestiodon species with the COI and CtyB “mammal c” primer sets may be the result of detection of a host bloodmeal from a species that is not represented in GenBank. While neither P. fasciatus nor P. skiltonianus are distributed in south Texas, other Plestiodon species such as P. obsoletus and P. tetragrammus are found in Dimmit and La Salle counties (Dixon, 2013). Sequences for these species for the targeted gene regions are not represented in GenBank.
A broad host range has been previously described for O. turicata, including both mammals and reptiles (Cooley and Kohls, 1944). Bloodmeal host detections previously described in cave-dwelling O. turicata in south-central Texas include raccoon, black vulture, and black-tailed rattlesnake DNA (Busselman et al., 2021). DNA from large mammal hosts, including domestic cattle and goats, white-tailed deer, and feral swine, has been identified in a small proportion of detected bloodmeals from O. turicata in other parts of south Texas in a recent study using metabarcoding (Balasubramanian et al., 2024). No large mammal species were represented in the detected bloodmeals in this study, despite the known presence of cattle, deer, feral swine, coyotes, bobcats, and African warthogs on the Chaparral Wildlife Management Area. Differences in habitat and environmental conditions as well as host species community composition may play a role in host selection of O. turicata.
Both the COI and CytB “Herp1/Herp2” primer sets amplified arthropod DNA in a total of 50 samples. The CytB “mammal c” primer set eliminated the amplification of arthropod DNA in all but two samples, but was prone to human contamination. No human detections with this primer set were confirmed with the other primer sets. Due to the settings in which ticks were collected, feeding on human hosts was considered unlikely. The variable numbers of detections of host bloodmeals with each of the three primer sets indicates that the use of multiple primer sets may help to improve host bloodmeal detection efforts when using Sanger sequencing; however, the amplification of tick DNA with these primer sets complicates analysis. Further primer optimization would be beneficial for future investigations of host ecology in soft ticks. The use of metabarcoding is another promising method that has been suggested to increase the potential for detection of bloodmeals in ticks from multiple host species (Balasubramanian et al., 2024), facilitating an even more detailed examination of host use.
Conclusions
The bloodmeal detections made in O. turicata from the Chaparral WMA indicate a broad host range, but suggest limited interactions with non-burrow-dwelling animals. This may be because of part to the xeric chaparral habitat, where tick activity may be limited by heat and low humidity to sheltering burrows. This may have implications for their capacity to act as vectors of introduced pathogens of concern and merits further investigation. The wide host range of O. turicata suggested herein may result in a dilution effect in areas of high host diversity on pathogens such as B. turicatae, which is thought to be acquired by ticks from burrowing rodents and wild canids (Armstrong et al., 2018; Nieto and Teglas, 2014). Further examination of the roles of host diversity and environment in soft tick host selection will help to improve our understanding of the transmission cycles of soft tick-associated pathogens and to better evaluate risks to human and domestic animal health.
The results of this study highlight the importance of further elucidating the identity and relationships of ticks identified as A. talaje in the United States, particularly given the detection of a potential pathogen of humans and companion animals in this species. Additional investigation into the identity and relationships between ticks morphologically identified as A. talaje and the associated relapsing fever group Borrelia is imperative to better understanding tick-borne disease risk in the southern U.S.
Footnotes
Acknowledgments
The authors acknowledge Texas Parks and Wildlife, Chaparral Wildlife Management Area, Lake Casa Blanca International State Park, and City of McAllen Parks and Recreation for permitting us to carry out sampling on state and city properties. The authors particularly thank Whitney Gann and Querubin Guerra for their onsite assistance. The authors acknowledge Spencer Hills for assistance with field sampling, Brian Rich, Taylor Donaldson, and Nadia Fernandez-Santos for aid with CO2 trap design, Sarah Hamer and Lisa Auckland for providing positive pathogen controls, and Jason Tidwell and Manuel Zavala for assistance with molecular work. The authors also acknowledge Pete Teel’s assistance with A. talaje identification and input on soft tick taxonomy. Sampling was carried out under Texas Parks and Wildlife State Park Scientific Study Permit 88-22, MOU N0-22-04802-W102.
Authors’ Contributions
S.E.M.M.—conceptualization, methodology, investigation, analysis, data curation, writing—original draft, writing—review and editing, project administration; L.P.M.—investigation and writing—review and editing, P.E.K.—conceptualization, methodology, writing—review and editing, supervision, and funding acquisition.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
This research was funded by Animal Health Project TEX09845.