
Abstract
Background:
Throughout the Americas, Lyssavirus rabies (RV) perpetuates as multiple variants among bat and mesocarnivore species. Interspecific RV spillover occurs on occasion, but clusters and viral host shifts are rare. The spillover and host shift of a big brown bat (Eptesicus fuscus) RV variant Ef-W1 into mesocarnivores was reported previously on several occasions during 2001–2009 in Flagstaff, Arizona, USA, and controlled through rabies vaccination of target wildlife. During autumn 2021, a new cluster of Ef-W1 RV cases infecting striped skunks (Mephitis mephitis) was detected from United States Department of Agriculture enhanced rabies surveillance in Flagstaff. The number of Ef-W1 RV spillover cases within a short timeframe suggested the potential for transmission between skunks and an emerging host shift.
Materials and Methods:
Whole and partial RV genomic sequencing was performed to evaluate the phylogenetic relationships of the 2021–2023 Ef-W1 cases infecting striped skunks with earlier outbreaks. Additionally, real-time reverse-transcriptase PCR (rtRT-PCR) was used to opportunistically compare viral RNA loads in brain and salivary gland tissues of naturally infected skunks.
Results:
Genomic RV sequencing revealed that the origin of the 2021–2023 epizootic of Ef-W1 RV was distinct from the multiple outbreaks detected from 2001–2009. Naturally infected skunks with the Ef-W1 RV showed greater viral RNA loads in the brain, but equivalent viral RNA loads in the mandibular salivary glands, compared to an opportunistic sample of skunks naturally infected with a South-Central skunk RV from northern Colorado, USA.
Conclusion:
Considering a high risk for onward transmission and spread of the Ef-W1 RV in Flagstaff, public outreach, enhanced rabies surveillance, and control efforts, focused on education, sample characterization, and vaccination, have been ongoing since 2021 to mitigate and prevent the spread and establishment of Ef-W1 RV in mesocarnivores.
Introduction
The process of viral emergence in a novel host involves multiple stages, but at minimum requires a productive spillover infection in the novel host and contact between the reservoir host and novel host or population (Childs et al 2007; Plowright et al 2017). Later stages of the process, which may be nonessential for emergence but serve as evidence of a host shift, include sustained viral transmission within the novel host in the absence of new spillover infections and potential viral adaptation to the novel host (Childs et al 2007; Plowright et al 2017). Ecological requirements for the emergence of zoonoses are often met through overlapping geographic distribution and spatial activity of reservoirs and novel hosts, whereas taxonomic diversity and relatedness are also strong modifying evolutionary factors impacting viral zoonotic risk (Mollentze and Streicker, 2020; Jacquot et al, 2022).
The history of Lyssavirus rabies (RV) evolution has repeatedly involved spillover and host shifts (Badrane and Tordo, 2001; Troupin et al, 2016). All mammals are susceptible to RV infection, in part because RV infects the nervous system via conserved receptor-binding domains, e.g., the nicotinic acetylcholine receptor (Jackson, 2020). Empirical studies of suspected wildlife RV host shift events have reported that selection upon the RV genome in a new host is transient and may not follow consistent nor predictable pathways of adaptation, with potential preadaptation of some RV variants to spillover (Kuzmin et al, 2012; Streicker et al, 2012; Borucki et al, 2013; Marston et al, 2017; Streicker and Biek, 2020). Throughout the Americas, RV is enzootic in multiple bat and mesocarnivore reservoir species (Gilbert, 2018), where transmission mostly occurs among conspecifics (Carey and McLean, 1983; Smith, 1989). It remains unclear why most RV variants are maintained by a single species reservoir in the Americas (Rupprecht et al, 2011). A mechanistic understanding of the host factors influencing this phenomenon has been lacking, yet a recent review synthesized hundreds of empirical studies and concluded that the relatedness between hosts may constrain RV pathobiology and can influence viral evolutionary adaptation and onward transmission in novel populations or species (Fisher et al, 2018; Mollentze et al, 2020).
Spillovers and host shifts of bat RV have been documented previously (Streicker et al, 2010; Streicker et al, 2012), yet the detection of spatiotemporal clusters of bat RV spillover in mesocarnivores has been rarely reported (Daoust et al, 1996). Unique evidence of multiple independent bat RV spillover events and host shifts into striped skunks (Mephitis mephitis) and gray foxes (Urocyon cinereoargenteus) in Flagstaff, Arizona, USA, was described during 2001–2009, associated with the big brown bat (Eptesicus fuscus) RV variant Ef-W1 (Leslie et al, 2006; Blanton et al, 2010; Kuzmin et al, 2012). While distinct outbreaks of Ef-W1 RV in mesocarnivores were reported during 2001, 2004–2005 and 2008–2009 in Arizona, only the 2001 and 2008–2009 cases formed independent monophyletic clusters (Kuzmin et al, 2012). Collaborative responses to the 2001–2009 outbreaks in Flagstaff, Arizona, were led jointly by Coconino County and the United States Department of Agriculture (USDA) involving enhanced rabies surveillance (ERS) and targeted wildlife rabies management to protect human and animal health (Slate et al, 2009).
On August 5, 2021, an RV spillover was confirmed in a striped skunk by the Wildlife Services program of the USDA,
Methods and Materials
Wildlife reported by the public or private organizations were submitted for RV diagnosis as part of ERS, by the USDA APHIS Wildlife Services and using the direct rapid immunohistochemical test (Patrick et al, 2019; Rupprecht et al, 2022), or for rabies public health surveillance by the Arizona Department of Health Services, Bureau of State Laboratory Services, Phoenix, Arizona, USA, using the direct fluorescent antibody test (Ronald et al, 2003). Brain samples from rabid animals were sent for typing and genomic characterization at either the Centers for Disease Control and Prevention (CDC), Poxvirus and Rabies Laboratory or the Wadsworth Rabies Laboratory of the New York State Department of Health. Carcasses from 15 rabid and two RV-negative striped skunks were shipped to the USDA APHIS Wildlife Services National Wildlife Research Center, Fort Collins, Colorado, USA, for postmortem tissue collection and real-time reverse-transcriptase polymerase chain reaction (rtRT-PCR) assay of brain and salivary gland tissues.
We opportunistically included parallel data and rtRT-PCR results from a pre-existing collaboration and archive of postmortem samples, comprised of 32 confirmed rabid striped skunks and seven RV-negative skunks that had been submitted for public health surveillance to the Colorado State University Veterinary Diagnostic Laboratory during 2018–2019. Sample RNA isolation methods are provided in the Supplementary Data S1.
Lyssavirus Rabies Typing and Genomic Sequencing
At Wadsworth laboratory, RV typing was performed using a modified triplex rtRT-PCR assay, amplifying two separate regions of the RV nucleoprotein (Nadin-Davis et al, 2009) and a beta-actin mRNA internal positive control (IPC; Wadhwa et al, 2017) on the Applied Biosystem 7500 FAST platform (Thermo Fisher Scientific, Waltham, MA, USA).
At the CDC laboratory, complete RV nucleoprotein and glycoprotein gene sequencing was performed as described in Dettinger et al (2022), except that RT and primary PCR was performed using SuperScript IV one-step reverse transcriptase-PCR system (Thermo Fisher, Waltham, MA, USA) following the manufacturer’s recommendations in 20 μL reactions. Sequencing was performed on the MinION using a flongle flow cell version FLG001 after library preparation with the LSK-109 ligation sequencing kit and EXP-PBC096 PCR barcoding kit (Oxford Nanopore Technologies, Oxford, UK). Basecalling was performed using guppy version 4.2.2 (Oxford Nanopore Technologies, Oxford, UK). Reference-based consensus sequences were generated using ivar 0.1 (https://github.com/andersen-lab/ivar) from mapping output of minimap2 v.2.16 (https://github.com/lh3/minimap2) and polished using medaka v1.0.1 (https://github.com/nanoporetech/medaka). Manual indel correction was then performed as described previously for the coding regions of the nucleoprotein and glycoprotein genes (Gigante et al, 2020). WGS was performed on cDNA generated from total RNA using Maxima H-minus double-stranded cDNA synthesis kit (Thermo Fisher, Waltham, MA, USA) after DNase digestion using ezDNase (Thermo Fisher, Waltham, MA, USA) following manufacturer’s protocols. cDNA was sequenced on a NovaSeq 6000 using the Illumina DNA prep kit following the manufacturer’s recommendations using half reagent volumes (Illumina, San Diego, CA, USA).
At the Wadsworth lab, WGS was performed as described previously with minor modifications (Brunt et al, 2020). Sample RNA eluates were evaluated for quality control by rtRT-PCR, then quantified by Nanodrop spectrophotometry (Thermo Fisher, Waltham, MA, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Total RNA was enriched using Poly(A) mRNA magnetic isolation module and NEBNext Ultra II Directional RNA Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA). Adapters were ligated with NEBNext Multiplex Oligos for Illumina, and libraries were quantified using the NEBNext Library Quant kit for Illumina (New England Biolabs, Ipswich, MA, USA). Samples were pooled and run on the Illumina NextSeq 500/550 using a Mid Output kit v2.5 (Illumina, San Diego, CA, USA).
Lyssavirus rabies quantification
We used a modified version of the pan-lyssavirus rtRT-PCR assay (Supplementary Figure S1.; Wadhwa et al, 2017) and compared relative viral loads based on cycle threshold values (Ct). The reactions were run on a BioRad CFX96 real-time PCR System using the AgPath-ID One-Step rtRT-PCR kit (Thermo Fisher, Waltham, MA, USA). Each reaction contained 5 μL of template RNA (diluted 1:10 in nuclease-free water), 3.5 μL of nuclease-free water, 12.5 μL 2× RT-PCR Buffer, 1 μL 25X RT-PCR Enzyme Mix, 0.5 μL each of 20 μM IPC β-actin forward and reverse primers and 20 μM LN34 forward and reverse primers, and 0.5 μL of 10 μM β-actin (HEX) and modified LN34 (FAM) probe. The thermocycling parameters were as follows: 30 min at 50°C, 10 min at 95°C, and 45 cycles of 1 s at 95°C, and 20 s at 56°C. Each sample was run in triplicate, and each PCR plate included a nontemplate negative control and RNA from a street RV (USDA Center for Veterinary Biologics, #92-5A) as a positive control. The rtRT-PCR results were evaluated using the BioRad CFX Maestro software package.
Phylogenetic and statistical analyses
The RV glycoprotein gene, protein, or genome sequences were aligned using MAFFT v.7.450 using the FFT-NS-I x1000 algorithm (Katoh et al, 2002; Katoh and Standley, 2013). Substitution models were determined based on LnL score from a model test performed in Mega7. Maximum clade credibility trees were estimated using BEAST v1.8.3 under GTR+G + I substitution model with two codon partitions (1 + 2 and 3), using an uncorrelated log normal relaxed clock and Bayesian skyline prior for the RV glycoprotein and WGS data. Additional clock and tree priors were tested. Tip dates were specified in years with a precision of 0.5 for the RV sequences analyzed (Supplementary Table S1).
An RV glycoprotein gene alignment containing representatives from major bat variants found in Arizona (Supplementary Table S2) was analyzed for evidence of selection in the Ef-W1 variant using branch-site models in the CODEML package in PAML 4.5 (Yang, 2007). Model A (includes codons under positive selection in the Ef-W1 variant, specified by NSsites = 2, model = 2, fixomega = 0) was compared to Model A1 (no positive selection allowed, specified by NSsites = 2, model = 2, fixomega = 1) by log likelihood test. Analyses were performed with a user tree generated using BEAST v1.8.3 under HKY + G + I substitution model, a strict clock and constant coalescent prior, CodonFreq = 2, and all ambiguous sites were removed. Sites where dN/dS > 1 were identified based on Bayes Empirical Bayes analysis using Pr(ω > 1) > 0.95 as a cutoff (Yang et al, 2005). Branches tested as the foreground lineage are depicted in Supplementary Figure S2.
Differences in rtRT-PCR detection of the two RV variants were evaluated for brain and salivary gland samples. The Ct values from both sample types were normalized using the 2-(ΔΔCt) approach (Livak and Schmittgen, 2001; Supplementary Figure S3.). We log transformed 2-(ΔΔCt) data to conform to a normal distribution based on the Shapiro–Wilk test (brain: W = 0.98, p = 0.41; salivary gland: W = 0.93, p = 0.16), and we tested for differences of transformed Ct value means between RV variants using Welch’s unequal variances t-test in R v4.1.3, evaluated with α = 0.05.
Results
A total of 22 cases of Ef-W1 RV variant was detected from striped skunks in northern Arizona during 2021–2023, in addition to one case in a gray fox during 2021 (Table 1, Fig. 1). Cases of Ef-W1 RV were detected from big brown bats in northern Arizona each year during 2021–2023, and one spillover infection was reported in a long-legged Myotis (Myotis volans) during 2022 (Table 1, Figs. 1 and 2). A single rabid ringtail (Bassariscus astutus) detected during October 2021 was infected with a Brazilian free-tailed bat (Tadarida brasiliensis) RV variant and one hooded skunk (Mephitis macroura) reported during September 2021 was infected with a South-Central skunk (SCSK) RV variant (Table 1, Fig. 1). Several other positive bats were detected in northern Arizona during 2023, infected with multiple bat RV variants (Table 1), including a novel bat RV described from New Mexico (Condori et al, 2022).
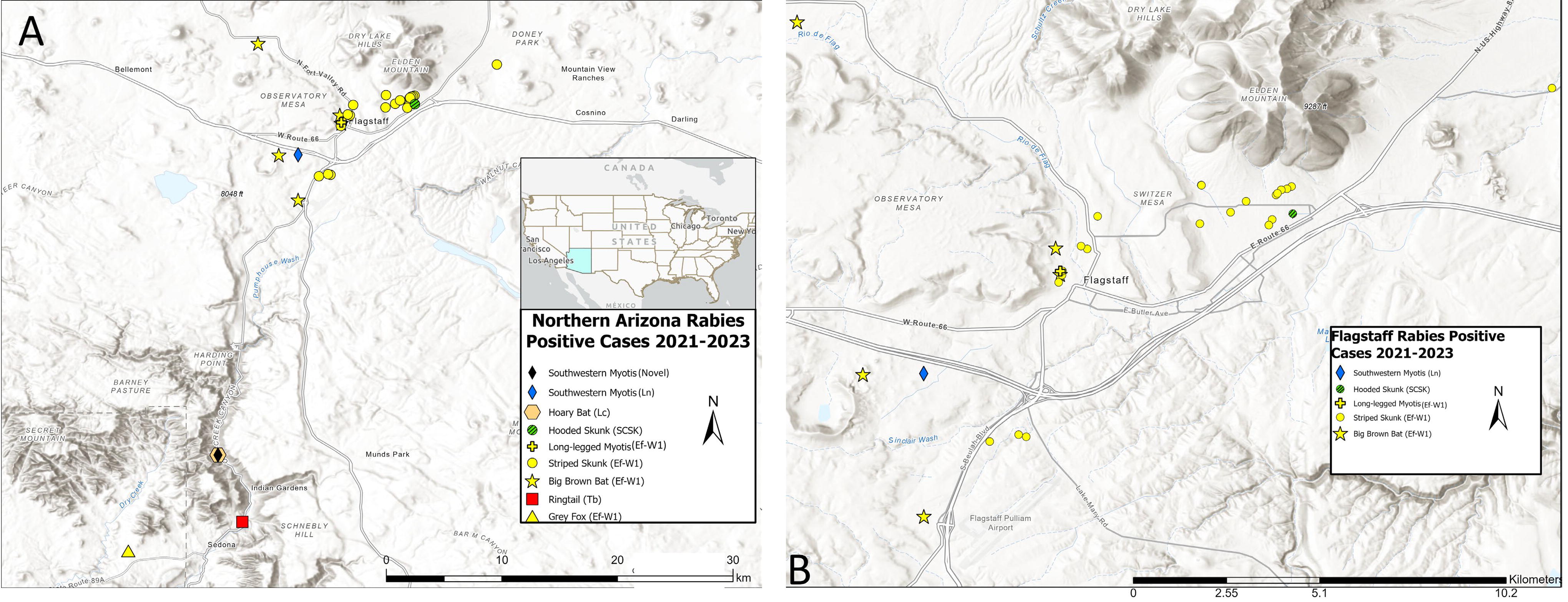
Locations, host species, and RV variant typing among rabid animal cases in northern Arizona during 2021–2023
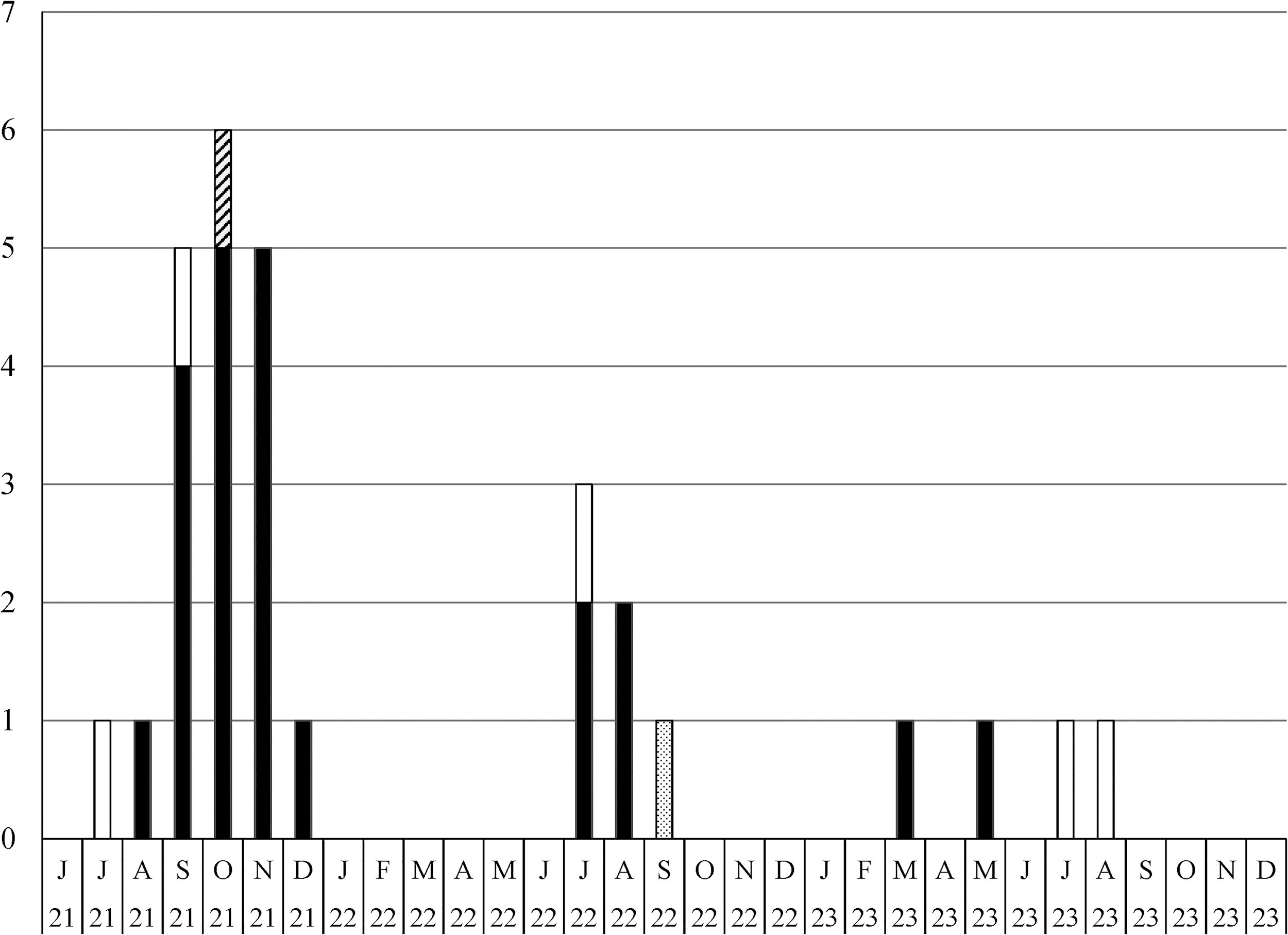
The prevalence of big brown bat (Ef-W1) Lyssavirus rabies variant in northern Arizona during 2021–2023, based on public health and enhanced rabies surveillance. Cases were detected from big brown bats (white bars), a long-legged Myotis bat (Myotis volans; dotted hatch bar), striped skunks (Mephitis mephitis, black bars), and a gray fox (Urocyon cinereoargenteus, diagonal hatch bar). Ef-W1, Eptesicus fuscus.
Rabid Bats and Mesocarnivores from Public Health and Enhanced Rabies Surveillance during 2021–2023 from Northern Arizona, USA. The RV Variant Typing Information is Described Where Available, or Clarified When Samples Were Not Typed (nt)
Two cases confirmed with Ef-W1 RV variant and two cases were not typed.
Novel bat RV variant as described in literature by Condori et al. (2022).
Ef-W1, Eptesicus fuscus; RV, Lyssavirus rabies.
The index case of Ef-W1 RV in striped skunks was detected in August 2021, while the greatest monthly incidence of Ef-W1 RV cases was documented in northern Arizona during September-November 2021 (Fig. 2). The majority of animals tested and rabid animals reported in northern Arizona during 2021–2023 were from USDA APHIS Wildlife Services ERS (Supplementary Table S3).
Phylogenetic reconstruction of relationships from Ef-W1 RV glycoprotein and WGS, including the prior outbreaks during 2001–2009, supported the independent emergence of the 2021–2023 outbreak (Figs. 3 and 4). The Ef-W1 sequences from skunks during 2021–2023 formed a monophyletic cluster that did not contain any of the Ef-W1 RV sequences isolated from bats during 2020–2023 (Figs. 3 and 4). The 2021–2023 outbreak of Ef-W1 in skunks was most closely related to sequences isolated from big brown bats in Arizona during 2001, 2010, and 2011 and an RV sequence from a cat (Felis catus) in Coconino County in 2005. The 2021–2023 Ef-W1 RV sequences from skunks shared a common ancestor with sequences from two Ef-W1 RV host shifts into skunks in 2001. However, 2001 and 2021–2023 skunk sequences form monophyletic clades in all phylogenies, separated by RV sequences collected from bats and other animals (Figs. 3 and 4). The 2009 Ef-W1 RV outbreak in gray foxes was more distantly related to both the 2001 and 2021–2023 outbreak clades.
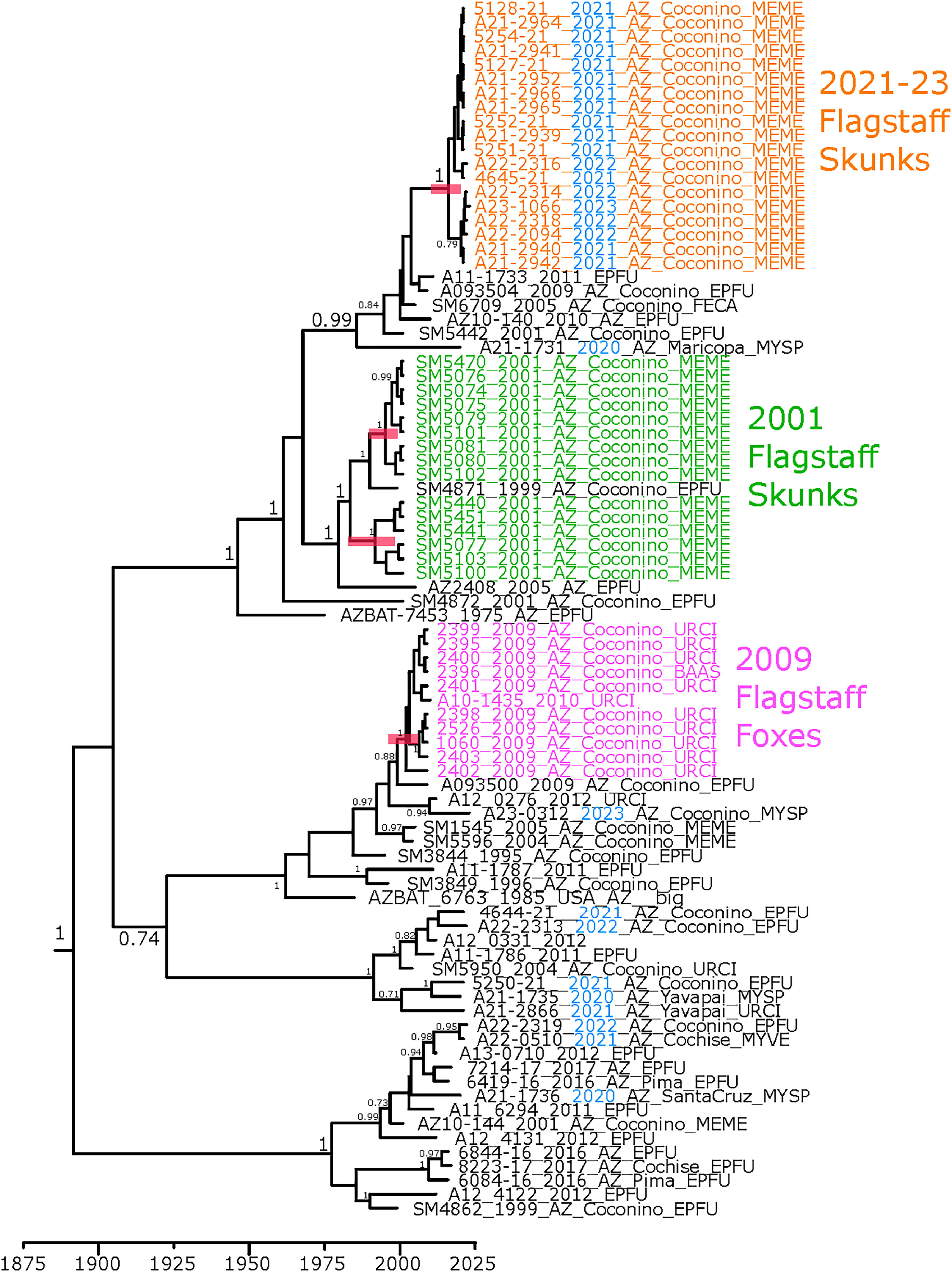
Phylogenetic analysis of Ef-W1 RV sequences from Arizona, USA. Phylogenetic tree estimated from alignment of RV glycoprotein gene sequences. Posterior support values >0.7 are displayed on the nodes. Scale is in years. Blue coloring indicates sample collected since 2020. Host shift clades are highlighted by colored sample names. Sample names follow the format ISOLATE_YEAR_STATE_COUNTY_HOST, and the RV variant JQ685942 (EF-W2) was used as an outgroup to root the tree. Ef-W1, Eptesicus fuscus; EPFU, Eptesicus fuscus; FECA, Felis catus; MEME, Mephitis mephitis; MYSP, Myotis species; MYVE, Myotis velifer; RV, Lyssavirus rabies; URCI, Urocyon cinereoargenteus.
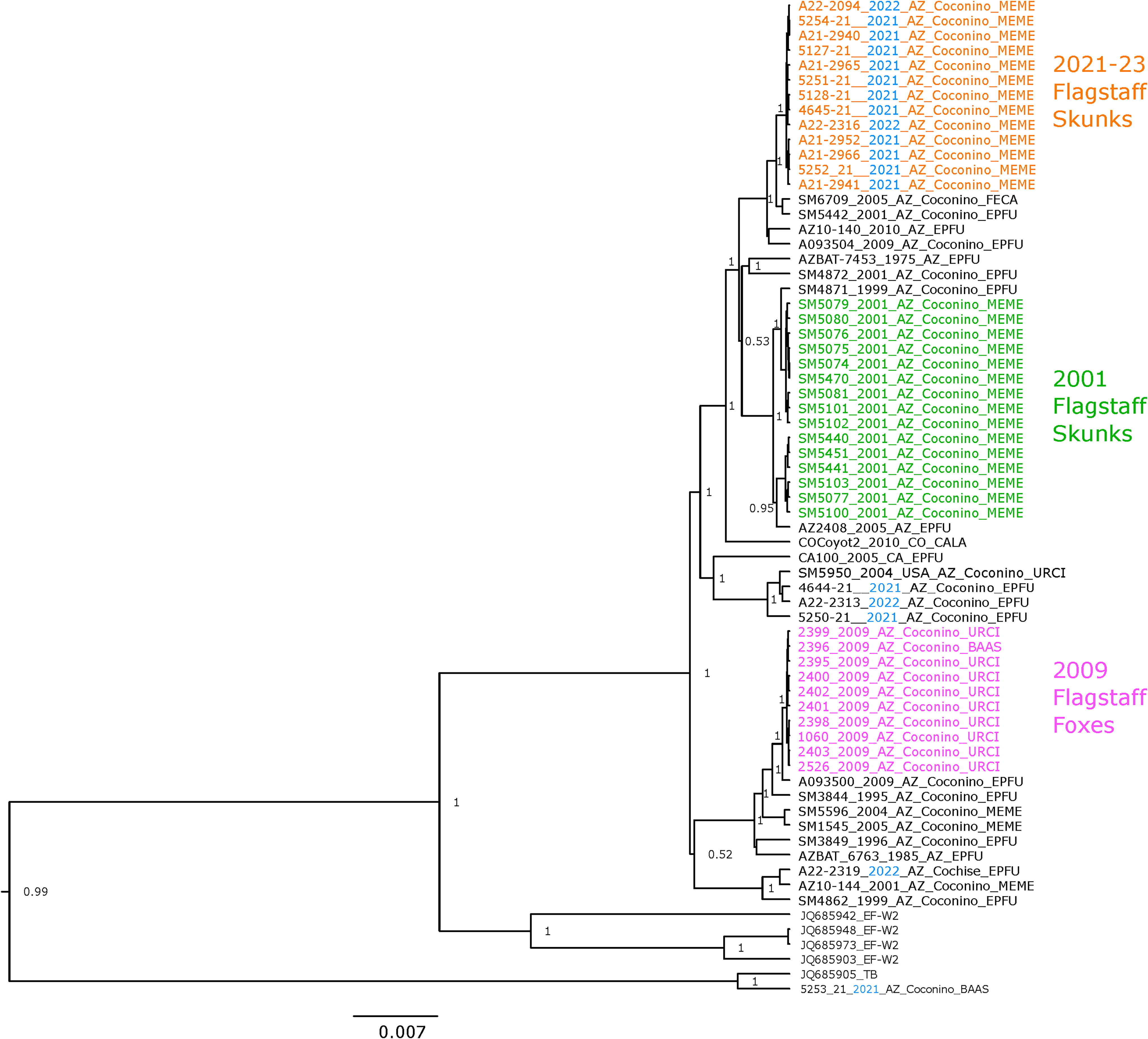
Phylogenetic analysis of RV WGS of Ef-W1 variant from Arizona and other states, USA. Posterior support values >0.7 are displayed on the nodes. Scale is in years. The tree was rooted using South-Central skunk RV variant. Host shift clades are highlighted by colored sample names. Sample names follow the format ISOLATE_YEAR_STATE_COUNTY_HOST. BAAS, Bassariscus astutus; CALA, Canis latrans; Ef-W1, Eptesicus fuscus; EPFU, Eptesicus fuscus; FECA, Felis catus; MEME, Mephitis mephitis; MYSP, Myotis species; MYVE, Myotis velifer; RV, Lyssavirus rabies; URCI, Urocyon cinereoargenteus; WGS, whole genome sequences.
Timing estimates for the most recent common ancestor (MRCA) of the 2021–2023 cases were 2015 or 2016, based on WGS and glycoprotein phylogenetic trees (Fig. 3, Table 2). The 95% highest posterior density (HPD) region for the MRCA based on the WGS included estimates as early as 2011 and as recent as 2019, suggesting the introduction of Ef-W1 into skunks likely occurred several years prior to detection in 2021. The MRCA of two sister clades of Ef-W1 documented from the 2001 outbreak in skunks from Flagstaff was estimated to be circulating prior to 1960, with MRCA of each sister clade estimated around 1995 (Table 2), in agreement with previous analyses that the two 2001 sister clades represented independent introductions (Kuzmin et al, 2012). The MRCA for the 2009 outbreak of Ef-W1 in gray foxes was estimated to occur during the early 2000s. In all four host shift events, MRCA estimations preceded detection in the new host by at least 2–3 years (Table 2). The MRCA time estimates reported provide further evidence of independent evolution of the prior and current Ef-W1 RV outbreaks in mesocarnivores from northern Arizona.
Age of MRCA and Mutation Rate Estimates from BEAST Analysis of Lyssavirus Rabies Whole Genome Sequences Shown in Figure 4. Several Parameters were Tested to Determine if Mutation Rate or Estimated Divergence Time Estimates were Influenced by Parameters Chosen. Mean and 95% HPD (in Brackets) are Shown. Estimates Produced from a Separate Alignment of 86 Glycoprotein Genes are also Shown
HPD, highest posterior density; MRCA, most recent common ancestor.
We examined glycoprotein sequences to investigate whether there was evidence of adaptation in the Ef-W1 RV variant, both within the four host shift clusters, and more broadly, across sequences. All glycoprotein sequences from the 2021 skunk Ef-W1 cases were identical (0 amino acid differences, 0–2 nucleotide differences) (Fig. 5), as observed within the 2009 fox, 2001a skunk, and 2001b skunk outbreak clades (Fig. 5). Within the Ef-W1 RV, two unique amino acid changes were observed in the 2021 clade of skunks (M462I and S496P), in comparison to the 2001–2009 outbreaks, yet these mutations were also observed among sequences from 2001, 2009, and 2010 big brown bats and a 2005 cat from Coconino County (Fig. 5). No unique amino acid changes were shared across all three spillover clusters relative to other Ef-W1 RV sequences, and no unique sites were under positive selection within the four host shift clusters, within the lineage containing all host shift clusters, or across all Ef-W1. Amino acids 272S, 403V, and 503S exhibited evidence of positive selection in both Ef-W1 and Ef-W2 compared with other bat RV variant lineages [P(ω > 1) = 0.702, 0.679, and 0.704, respectively]. Whether these amino acid changes preadapt the virus for a host shift is not clear, as no host shifts have been reported for variant Ef-W2.
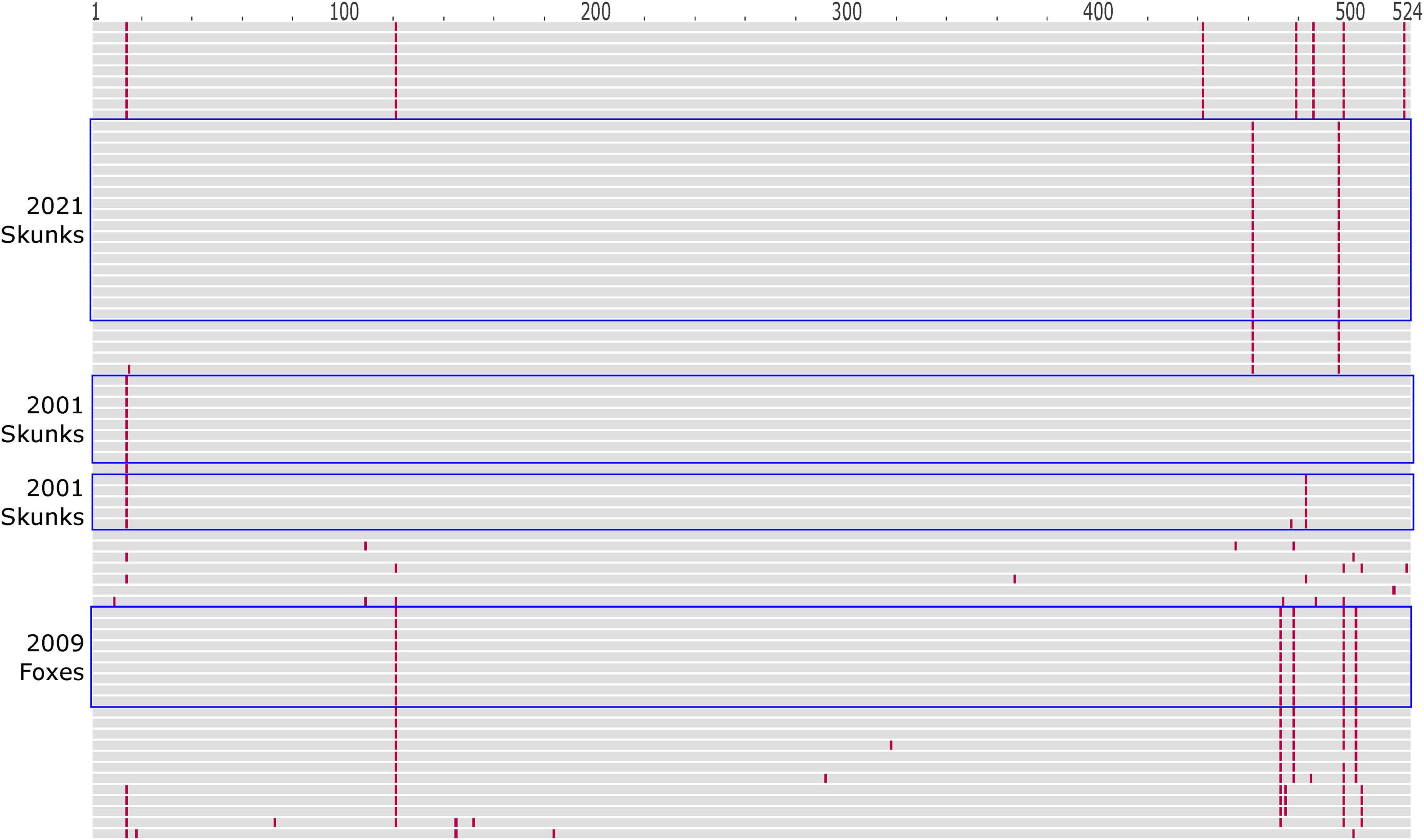
Amino acid changes in the glycoprotein among Ef-W1 RV variant sequences shown in Figure 3. Each horizontal line represents one protein sequence. Amino acids identical to the RV glycoprotein sequence from GenBank accession JQ685956.1 (Arizona bat 1975) are shown in gray. Amino acid changes relative to JQ685956.1 are shown in red. Sequences from 2021, 2009, and 2001 Ef-W1 RV mesocarnivore host shift clusters are identified with blue boxes. Ef-W1, Eptesicus fuscus; RV, Lyssavirus rabies.
We detected RV by rtRT-PCR in all brain samples from rabid animals, whereas no RV was detected in brain samples from skunks with a negative rabies diagnosis (Table 3, Supplementary Table S4; skunks with negative RV diagnosis not shown). The mean RV Ct was 17.8 (range: 16.4–21.7) for the brains of 15 Ef-W1-positive skunks (Table 3) and 17.1 (range: 14.4–21.5) for the brains of 32 SCSK-positive skunks (Supplementary Table S4). A two-sided t-test for differences in mean 2-(ΔΔCt) brain Ct values between rabid striped skunks infected with Ef-W1 or SCSK RV variant(s) suggests a marginal difference (t = 1.94, df = 19.2, p = 0.07; Supplementary Figure S4).
The Mean Cycle Threshold Values (Ct) Across Real-Time Reverse Transcriptase PCR Triplicate Runs for the Detection of a Beta-Actin IPC and Lyssavirus rabies RNA (mLN34) in Brain and Mandibular SG Tissues of Skunks Naturally Infected with Big Brown Bat (Eptesicus fuscus) Lyssavirus rabies Variant (Ef-W1) in Coconino County during 2021–2023
IPC value considered invalid test for RV detection, scored as indeterminate.
Ef-W1, Eptesicus fuscus; IPC, internal positive control; RV, Lyssavirus rabies; SG, salivary gland.
We encountered rtRT-PCR RV detection curves with atypical morphology from some salivary gland samples. Samples with atypical RV detection curves were retested with additional RNA (from 5 μL to 8.5 μL), yet the detection curve morphology remained atypical. The salivary gland samples with atypical RV detection curves were scored as indeterminate for quantification, comprising two (13% of 15) Ef-W1 RV samples and six (19% of 32) SCSK RV samples. We detected RV in 9 of 12 (75%) mandibular salivary glands from rabid skunks infected with Ef-W1, with a mean Ct of 23.7 (range: 17.0–32.1; Table 3), and from 9 of 26 (34%) mandibular salivary glands from rabid skunks infected with SCSK variant, with a mean Ct of 26.4 (range: 18.4–35.0; Supplementary Table S4). The IPC failed for one rabid skunk mandibular salivary gland sample infected with Ef-W1 RV variant, with RV detection scored as indeterminate (Table 3). A two-sided t-test for differences in mean 2-(ΔΔCt) mandibular salivary gland Ct values between rabid striped skunks infected with Ef-W1 or SCSK RV variant(s) was not significant (t = 1.56, df = 13.7, p = 0.14; Supplementary Figure S5).
From nine skunks where Ef-W1 RV was detected in mandibular salivary glands, eight had sublingual gland tissue available for rtRT-PCR testing and seven of the eight skunks also had RV detected in sublingual salivary glands (Supplementary Table S5). In contrast, the rtRT-PCR test of mandibular salivary glands for one skunk (Ef-W1, AZ011; Table 3) failed on the beta-actin IPC, yet RV was detected from sublingual salivary glands (Supplementary Table S5). Parotid salivary gland tissues from 15 skunks infected with Ef-W1 were tested by rtRT-PCR, but samples from three skunks were excluded due to atypical RV detection curves and indeterminate results, leaving five of 12 (42%) skunks with RV detected from the parotid salivary glands (i.e., four skunks with RV positive and one skunk with indeterminate mandibular gland results; Supplementary Table S5). Sublingual and parotid gland tissues from rabid skunks of northern Colorado, infected with SCSK RV, were not available for comparison.
Discussion
The reemergence of the Ef-W1 big brown bat RV in mesocarnivores from northern Arizona, USA, during 2021–2023 strengthens the evidence suggesting that this RV variant may be preadapted for spillover infections and host shifts (Kuzmin et al, 2012). The 2021–2023 outbreak in skunks shared a MRCA with singular detections of Ef-W1 spillover in a cat from 2005 and circulation of RV in big brown bats during 2009–2011, but the 2021–2023 outbreak was distinct from the clade(s) of Ef-W1 viruses that emerged in mesocarnivores in northern Arizona during 2001–2009. Results of this study suggest a high potential for onward transmission within striped skunk populations in this region. Salivary shedding of RV is a prerequisite for transmission by bite, yet RV excretion in the salivary glands of rabid skunks may be a transient phenomenon, depending on variation in RV dissemination from the central nervous system to and replication in peripheral tissues (Charlton et al, 1983; Charlton et al, 1987).
Regional studies of skunk RV variant epizootiology report an overwhelming majority of cases occurring in striped skunks (Dragoo et al, 2004; Oertli et al, 2009; Pepin et al, 2017). Given the context of the prior emergence of Ef-W1 RV in striped skunks and gray foxes in northern Arizona during 2001–2009 and nearly exclusive reporting of 2021–2023 cases in striped skunks, there is a valid reason for ERS targeting mesocarnivores in the region, although onward transmission may be more likely in striped skunks. An emerging RV variant in mesocarnivores is concerning to managers due to the known challenges of targeting skunks with cost-efficient control strategies, as reviewed elsewhere (Slate et al, 2009; Wohlers et al, 2018).
The close phylogenetic relationship of the cases during 2021–2023 suggests evidence of onward transmission within skunks. The level of RV RNA copies in the brains of naturally infected skunks trended toward higher viral RNA loads for the emerging Ef-W1 RV compared to a skunk-adapted SCSK RV, yet there was also greater variability due to a smaller number of Ef-W1 RV infected skunks sampled. While the RV RNA copy levels from naturally infected skunk mandibular SGs were indistinguishable between the two RV variants, not all glands could be evaluated quantitatively in the comparison due to the presence of indeterminate results. Marston et al (2017) suggested that increased RV population genomic diversity within spillover hosts could result in a longer incubation time allowing viral loads and diversity to accumulate, increasing the chances for highly adapted genotypes to be transmitted to conspecifics. The trend toward greater Ef-W1 RV RNA in skunk brains, also supported by equivalent amounts of RV RNA in the salivary glands, may indicate either a high replication efficiency or a longer incubation period in the spillover host, despite our lack of evidence for positive selection on the reemerging Ef-W1 variant. One limitation with the salivary gland results may be individual variation among skunks in the disease progression at the time of submission for public health or ERS surveillance, and caution may also be warranted regarding the inference to RV shedding and transmission risk from salivary glands, given greater rates of RV isolation previously reported from the salivary glands of naturally infected skunks in northern Colorado (Jimenez et al, 2019). Future comparisons between rtRT-PCR and RV isolation from salivary glands, for contextualizing RV shedding potential, are recommended, and deep sequencing of salivary glands may lend insight to the processes of RV evolution and novel host adaptation. Campaigns to enhance public awareness, education, outreach, and enhanced surveillance activities are warranted in the southwestern USA, for timely detection and response to emerging and reemerging bat RV variants in mesocarnivores, with impacts for human and animal health.
Footnotes
Acknowledgments
The authors thank T. Spraker and M. Selleck for assistance with the samples from northern Colorado. The authors also thank A. Barbee for technical assistance with the sample curation, processing, and data collection.
Authors’ Contribution
All authors contributed substantively to the report, and all authors approve of the findings described therein.
Author Disclosure Statement
The authors have no conflicts of interest to declare with respect to the contents or findings reported. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention, the US Department of Agriculture, nor any partnering agencies.
Funding Information
This research was supported by the US Department of Agriculture, Wildlife Services, National Rabies Management Program.
Supplementary Material
Supplementary Data S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.