
Abstract
Francisella tularensis is the etiologic agent of tularemia, a bacterial zoonotic disease. The genome of F. tularensis shows a recent evolutionary change, especially in reservoirs. Variable number of tandem repeats (VNTR) is described as a high-speed molecular clock and can thus be used as a high-resolution typing system. The main objective of our study was to investigate the molecular diversity of F. tularensis strains and reveal possible sources of infection. Using real-time PCR targeting the ISFtu2 region, we successfully amplified targeted DNA in 13/31 Slovenian patients with a clinical diagnosis of tularemia, and with PCR targeting the fopA gene, we obtained 11/13 PCR products. Sequencing revealed that all samples were identified as F. tularensis subsp. holarctica. We successfully obtained one F. tularensis isolate from a lymph node aspirate by culture on chocolate agar. Our isolate was clustered into major clade B12 (subclade B43). We optimized VNTR typing to be used directly on clinical samples. Multiple-locus VNTR analysis (MLVA) revealed five unique MLVA types; 45.5% samples had the same MLVA type, another 27.3% shared a different MLVA type, and each of the remaining had a unique MLVA type. Most samples differed at only two VNTR markers (Ft-M03 and Ft-M06). Additionally, we investigated samples from small mammals (n = 532) and Ixodes ricinus ticks (n = 232) captured in the same geographical area in which patients with tularemia were found. No F. tularensis DNA was detected in samples of small mammals or I. ricinus ticks. The diversity of MLVA types in Slovenia was high, despite the small region, but most of the samples from the same region shared the same MLVA type. Our results suggest that MLVA is a useful tool for quick molecular characterization of F. tularensis directly from patient samples, especially when investigating geographically localized outbreaks.
Introduction
F
The case rate of tularemia in Europe is stable, with occasional outbreaks, including in Czech Republic, Kosovo, Bulgaria, Germany, Sweden, Finland, Spain, Turkey, France, and Norway (Carvalho et al. 2014). Slovenia is situated in Central Europe and has only rare and sporadic cases of tularemia. In the last 20 years, up to two cases per year have been reported (ECDC 2019). However, between 2012 and 2013, six patients with the ulceroglandular form of tularemia were recognized, all residing in or visiting the same town (Rojko et al. 2016). Epidemiological data indicated transmission by a tick bite in 50% of patients (Rojko et al. 2016). Ixodes ricinus is the most prevalent species of ixodid ticks in Slovenia. Dermacentor reticulatus is present only in the northeastern part of the country from where most tularemia cases were reported before 2012.
There are four recognized subspecies of F. tularensis: tularensis, holarctica, mediastatica, and novicida (Sjöstedt 2003). F. tularensis subsp. holarctica is present in most of the northern hemisphere (Vogler et al. 2009a). Subspecies identification is clinically important since different subspecies have different case fatality rates (Leelaporn et al. 2008, Carvalho et al. 2014). Nevertheless, because of the need for a high-containment laboratory due to the risk of laboratory-acquired infections, culture isolation and biochemical typing are often avoided (Petersen and Schriefer 2005, CDC 2015).
Molecular typing assays such as pulse field gel electrophoresis, whole-genome sequencing (WGS), multiple-locus variable number of tandem repeat (VNTR) analysis (MLVA), insertion–deletion markers (INDELs), and single-nucleotide polymorphisms (SNPs) allow safe characterization directly from isolated nucleic acids (Farlow et al. 2001, Garcia Del Blanco et al. 2002, Broekhuijsen et al. 2003, Johansson et al. 2004, Staples et al. 2006, Larsson et al. 2007, Vogler et al. 2009b, Gyuranecz et al. 2012). Strains of different geographical origin have highly conserved genomic sequences (Sjöstedt 2003). With WGS analysis, several genome sequences for Francisella spp. have become available and our understanding of differences among species, gene order, subspecies, and genetic subpopulations has advanced (Larsson et al. 2009, Johansson et al. 2010).
VNTR is described as a high-speed molecular clock (van Belkum 1999) and can thus be used as a high-resolution typing system (Johansson et al. 2004, Wahab et al. 2014). The genome of F. tularensis has shown a recent evolutionary change, especially in reservoirs (Larsson et al. 2009). Likewise, molecular typing methods can be used to investigate natural reservoirs and transmission routes (Fujita et al. 2013).
The main objective of our study was to investigate the genetic diversity of F. tularensis strains in Slovenian patients and to reveal possible sources of infection. Since culturing Francisella spp. is hazardous and tedious work and WGS directly from a patient's sample is still very expensive (Johansson et al., 2010), we decided to explore the possibility of utilizing MLVA for investigation of the molecular characteristics of Slovenian isolates. Additionally, tularemia may occur in a small, restricted geographical area, where cases may appear annually, but can later be absent for more than a decade. The reasons for such temporal and spatial variations are not yet understood. The aim of the study was thus to investigate the genetic diversity of tularemia in Slovenian patients to better understand this phenomenon.
Materials and Methods
In a 14-year period (2004–2018), 31 patients with a clinical diagnosis of tularemia were treated in Slovenia. The clinical diagnosis was microbiologically confirmed either with detection of IgM/IgG antibodies by commercial indirect immunofluorescence assay (Francisella/Brucella 12-well MIF Substrate Slides; Fuller Laboratories) or with real-time PCR (Versage et al. 2003). Before 2012, most of the tularemia cases were diagnosed in the northeastern part of the country, but in 2012 and 2013, a cluster of six cases appeared in the central part of Slovenia (Fig. 1) (Rojko et al. 2016).
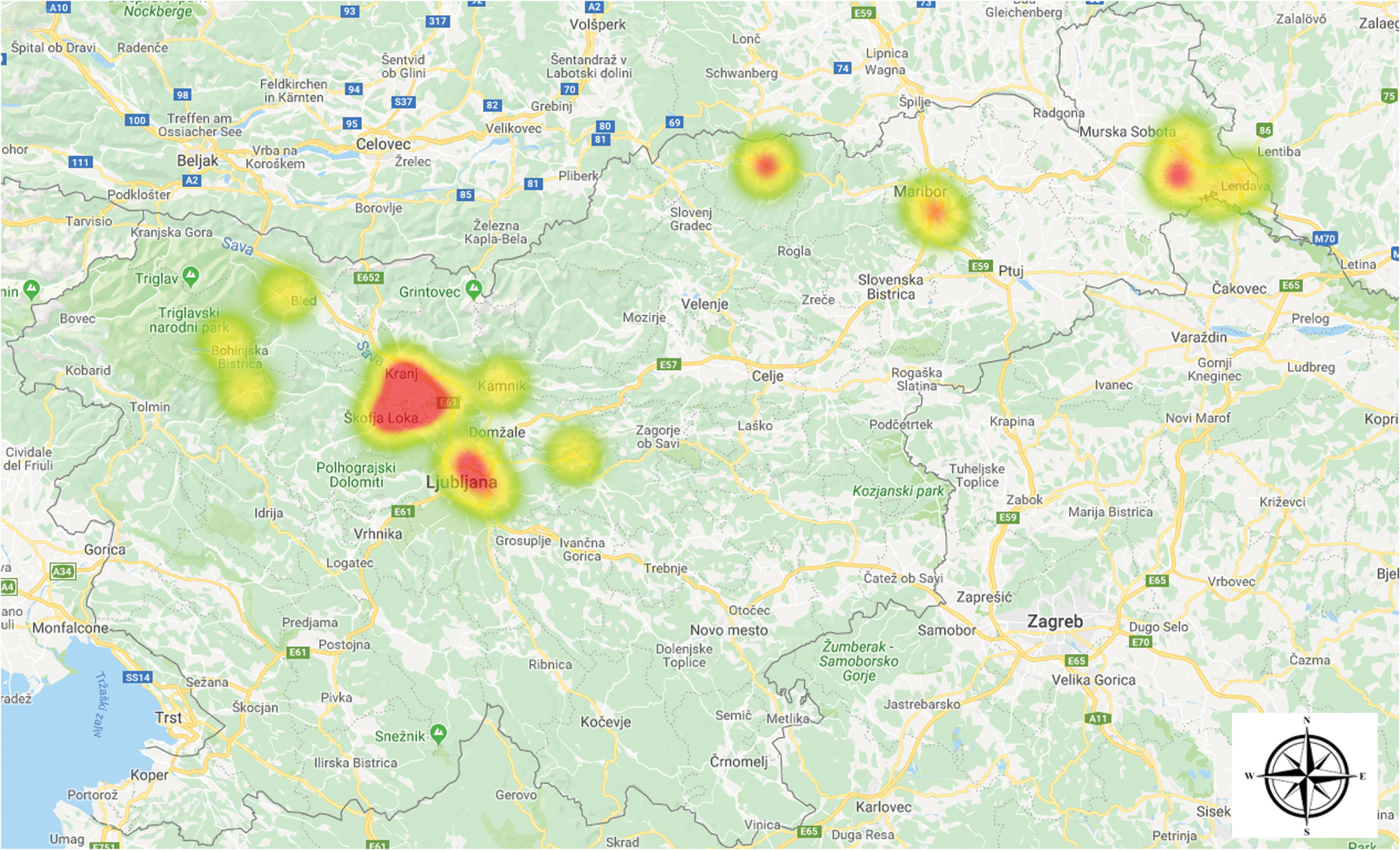
Clusters of human tularemia cases in Slovenia based on patients' place of residence. Color images are available online.
We additionally investigated samples from small mammals (Apodemus spp. [n = 358], Crocidura spp. [n = 5], Mus spp. [n = 8], Myodes spp. [n = 115], Microtus spp. [n = 31], Neomys spp. [n = 7], and Sorex spp. [n = 8]) and I. ricinus ticks (n = 232) captured in the same geographical area in which patients with tularemia had been found.
Human samples were sent to the Faculty of Medicine, Institute of Microbiology and Immunology, for diagnostic purposes. All procedures involving animals were approved by the National Ethics Committee and the Administration of the Republic of Slovenia for Food Safety, Veterinary and Plant Protection (permit no. U34401-15/2018/8, 18.7.2018). Animal care and treatment were conducted in accordance with institutional guidelines and international laws and policies (Directive 2010/63/EU on the protection of animals used for scientific purposes).
We extracted DNA from patients' blood, wound ulcer swab, or lymph node aspirate samples; rodent samples; and ticks using a DNA Mini Kit (Qiagen, Germany) and amplified tularemia DNA with real-time PCR targeting the ISFtu2 region (Versage et al. 2003). For determination of F. tularensis subspecies, we amplified the fopA gene with nested PCR (Fulop et al. 1996). Additionally, in the BSL-3 laboratory, we cultivated/inoculated a lymph node aspirate from patient no. 9 (described in detail by Rojko et al. 2016) on chocolate agar.
Whole genome was sequenced using an Ion 316 chip loaded into an Ion Torrent PGM benchtop sequencer (Applied Biosystems). Library preparation using 1 μg of extracted DNA was performed with an Ion Xpress Fragment Library Kit (Life Technologies). For size selection, the E-gel system was used according to the manufacturer's instructions. All steps were controlled by Caliper LabChip GX using an HT DNA High Sensitivity Assay Kit (Caliper Life Sciences, Germany). Template preparation was performed with a OneTouch 2 instrument and an Ion PGM Template OT2 200 Kit (Life Technologies) according to the manufacturer's instructions.
We selected six VNTR markers (Ft-M03, Ft-M06, Ft-M22, Ft-M24, Ft-M20, and Ft-M21) with the highest variability on a subspecies level (Johansson et al. 2004, Wahab et al. 2014) and performed MLVA directly from real-time PCR-positive clinical material. The VNTR markers, Ft-M03, Ft-M06, Ft-M22, Ft-M24 (Vogler et al. 2009b), Ft-M20, and Ft-M21 (Johansson et al. 2004), were amplified as follows: 1 × PCR supermix (Invitrogen), 2.5 μL of template DNA, at different concentrations of forward and reverse primers (Table 1), in a total volume of 25 μL. The reaction mixtures were incubated at 94°C for 2 min and then cycled at 94°C for 15 s, 60°C for 30 s, and 72°C for 40 s for 45 cycles, with a final incubation at 72°C for 5 min. Primers used were obtained from Applied Biosystems (Johansson et al. 2004, Vogler et al. 2009b). Forward primers were labeled with fluorescent dyes (Table 1).
Multiple-Locus Variable Number of Tandem Repeat Analysis Primer Sequences, Fluorescent Labels, and Final Concentrations
Amplicons were pooled into two combinations; (a) Ft-M03 (6 μL), Ft-M06 (5 μL), and Ft-M20 (3 μL) and (b) Ft-M21 (3 μL), Ft-M22 (3 μL), and Ft-M24 (10 μL) and diluted 20 times. Two microliters of each pool was analyzed with capillary electrophoresis (API 3500 Genetic Analyzer), POP7 polymer, and GeneScan™-1200 ROX™ size standards (Applied Biosystems). GeneMapper (Applied Biosystems) software was used to determine the size of amplicons and to calculate the number of repeats at each VNTR marker.
Results
Using real-time PCR targeting the ISFtu2 region, we successfully amplified targeted DNA in 13/31 samples, with Ct values ranging from 19.5 to 31.9 (Table 2).
Multiple-Locus Variable Number of Tandem Repeat Analysis Results in Conjunction with Date of Case Recognition, Location, Sample Type, Subspecies Identification, and Real-Time PCR Cycle Threshold Values
Newly found repeat number in this locus.
Described in detail by Rojko et al. (2016).
Isolation in bacterial culture.
MLVA, multiple-locus variable number of tandem repeat analysis; NS, not successful.
Using PCR targeting the fopA gene, we obtained 11/13 PCR products that were all subsequently sequenced, and the sequences revealed that all samples belonged to F. tularensis subsp. holarctica. Both samples in which subspecies determination was unsuccessful had a low bacterial load (Ct value >30.0).
We successfully obtained one F. tularensis isolate from a lymph node aspirate by culture on chocolate agar, from patient no. 9. Small, light gray, reflective, soft, and easily emulsified colonies were observed 3 days after inoculation. The biochemical characteristics were catalase—weakly positive, oxidase—negative, H2S—positive, and acid produced from glucose and maltose, but not from sucrose. Resistance to erythromycin was observed with antimicrobial testing.
Whole genome (WGS) from successfully cultured F. tularensis was sequenced. After multiple alignments (mapping to the reference genome and de novo assembly), we obtained 1830511 base pairs, which covered 96.5% of the reference genome LVS (NC_007880). Sequencing data have been submitted to the European Nucleotide Archive (study accession no. PRJEB34471). Gained reads were downsampled using in-house Python scripts to an estimated 40-fold coverage. Phylogenetic analysis based on WGS confirmed that the isolate is closely related to LVS (NZ_CP009694) and LVS (NC_007880) strains isolated in USA and the FSC200 (NC 019551) strain isolated in Sweden. By comparing conserved canonical SNPs, our isolate was clustered into major clade B12 (subclade B43), together with isolates from Central–Eastern Europe (Koene et al. 2019).
Analysis of isolates from Slovenian patients, combining all six VNTR markers, revealed that most samples differed at only two VNTR markers (Ft-M03 and Ft-M06), except in one sample in which different repeat numbers were also found at markers Ft-M22 and Ft-M24 (Table 1). In comparison with previous studies (Johansson et al. 2004, Wahab et al. 2014), we found new repeat numbers at three loci: Ft-M03, Ft-M20, and Ft-M21. Five samples (45.5%) had the same MLVA type (6,5,2,2,4,2), another three (27.3%) shared a different MLVA type (8,4,2,2,4,2), and each of the remaining had a unique MLVA type (Table 1).
Comparing the geographical area of patients' residence with MLVA type revealed that not all patients residing in the same area shared the same MLVA type (Fig. 2), which might suggest a different infection source. No F. tularensis DNA was detected in samples from small mammals and I. ricinus ticks.
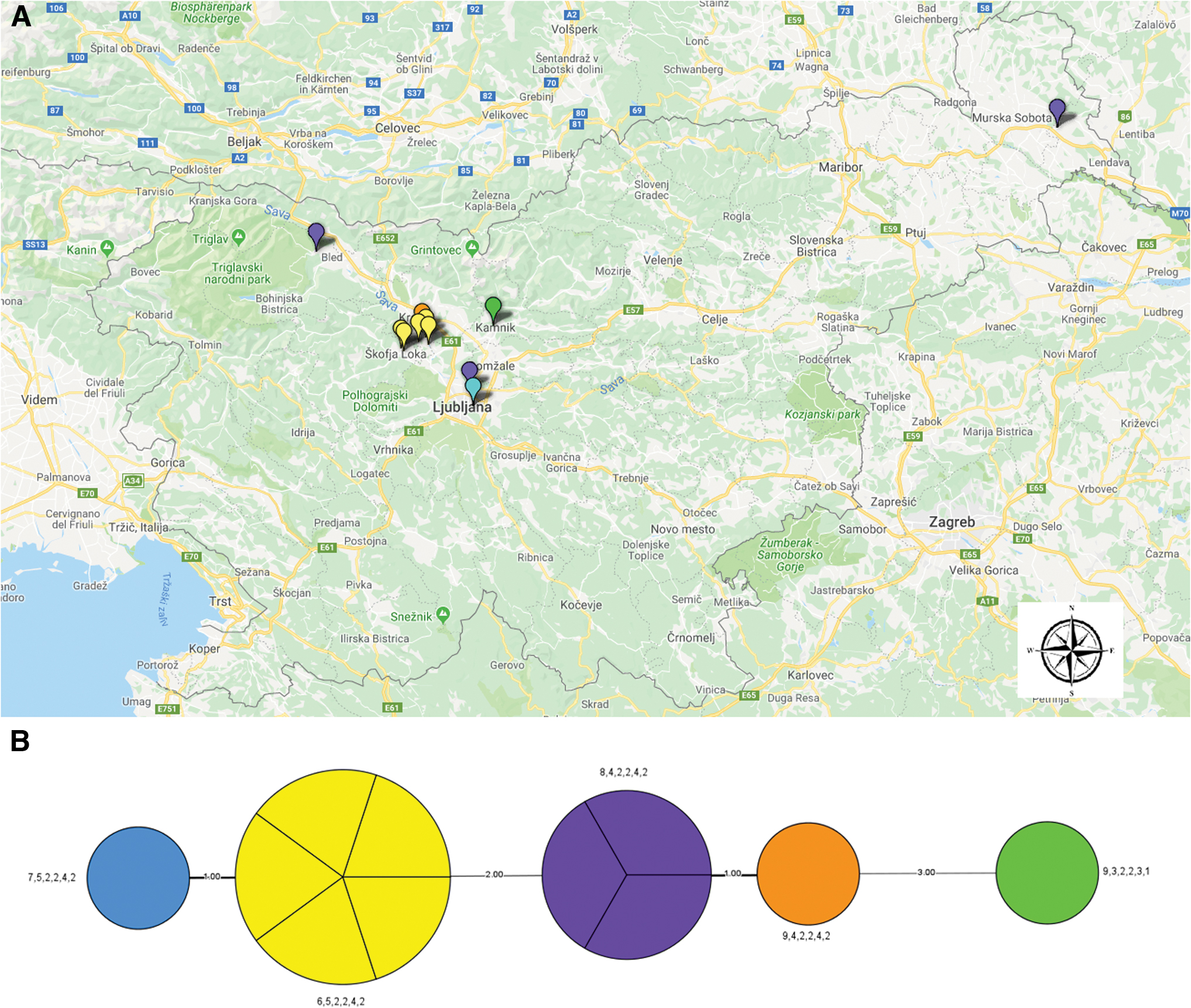
Distribution of MLVA types in Slovenia.
Discussion
In Slovenia, tularemia cases are sporadic; in the last 20 years, up to two cases were reported per year (ECDC 2019), but the numbers have recently been rising. Moreover, new geographical areas seem to be affected; in 2012, the first clinical cases were also diagnosed in the western part of the country (Rojko et al. 2016). In light of these recent events and since the disease is still neglected in Slovenia, the aim of the study was to investigate the molecular diversity of F. tularensis strains and possibly reveal the source of infection. Since the disease is only rarely recognized by primary care medical doctors, in most patients, the time to correct diagnosis is several weeks (9) and serology is thus most commonly used for microbiological confirmation of clinical suspicion. Real-time PCR is used mainly for detection of bacterial DNA in lymph node aspirates and ulcer swabs.
In our study, we successfully amplified F. tularensis DNA in 41.9% of investigated samples, and lower Ct values were detected in lymph node aspirates than in blood samples. With sequencing of the fopA gene, we confirmed that all patients were infected with F. tularensis subsp. holarctica, as expected, since this subspecies is the most prevalent in Europe and in most of the northern hemisphere (Vogler et al. 2009a). WGS of the cultivated isolate revealed that it belongs to major clade B12 and subclade B43. Strains belonging to major clade B12 are predominantly found in several countries of Central–Eastern Europe and in isolates from Czech Republic, Finland, Georgia, Russia, Slovakia, and Ukraine (Chanturia et al. 2011, Gyuranecz et al. 2012).
Since it was confirmed that all our samples are subspecies holarctica, we selected six VNTR markers (Johansson et al. 2004) to further investigate the molecular diversity of F. tularensis in patients' samples. MLVA is usually done on isolated bacterial cultures, but since we did not have fresh material to attempt culture isolation, we tried to optimize VNTR typing directly from clinical samples. We were able to analyze clinical samples with higher DNA loads, up to Ct 28. We successfully analyzed 84.6% of PCR-positive samples and discovered five unique MLVA types and two clusters. Most variations were found at the Ft-M03 and Ft-M06 loci, which are the most rapidly mutating markers (Johansson et al. 2004, Vogler et al. 2009b, 2011, Wahab et al. 2014).
In comparison with previous studies (Johansson et al. 2004), we found a new repeat number in three loci, Ft-M03, Ft-M20, and Ft-M21. The diversity of MLVA types was observed to be high not only in Slovenia, but also in other European countries (Vogler et al. 2009a, 2011). Most of the samples (5/6) from the same region (Mavčiče/Škofja Loka) shared the same MLVA type (6,5,2,2,4,2), which suggests the same source of infection. However, the MLVA type (8,4,2,2,4,2) was confirmed in patients residing in very distant regions (Fig. 2). Due to the limited number of tularemia cases in Slovenia in general, the low number of samples encompassed in this article is a limitation of the study.
However, our results suggest that MLVA is a useful tool for quick molecular characterization of F. tularensis directly from patient samples, especially when investigating geographically localized outbreaks. Our geographical analysis was based on patients' place of residence since studies on other vector-borne pathogens in Slovenia have indicated that most infections are locally acquired (Fajs et al. 2012, Korva et al. 2013), although there is always a possibility of acquiring the infection in other areas since tick bites can often be overlooked. To investigate a possible source of infection, we analyzed rodents and ticks captured in locations in which tularemia patients originated.
No F. tularensis DNA was detected in ticks' or rodents' tissue samples, but the number of investigated Microtus spp., which are probably the primary reservoirs of F. tularensis, was low (5.8%) (Duzlu et al. 2016). Nevertheless, since monitoring of vectors started in 2014, a year after detection of the cluster of cases in Mavčiče, this might also be due to interannual changes in the vector population, and a study covering a longer period might give different results.
Conclusions
Using sequencing of the fopA gene, we confirmed that all patients were infected with F. tularensis subsp. holarctica. WGS of the cultivated isolate revealed that it belongs to major clade B12 and subclade B43. Using VNTR, we were able to analyze clinical samples with higher DNA loads, up to Ct 28. We discovered 5 unique MLVA types and two clusters. Most variations were found at the Ft-M03 and Ft-M06 loci, and we found a new repeat number in three loci, Ft-M03, Ft-M20, and Ft-M21. The diversity of MLVA types in Slovenia was high, although most of the samples from the same region (Mavčiče/Škofja Loka) shared the same MLVA type. These facts lead to the conclusion that MLVA might also be useful for monitoring genetic variants of F. tularensis in different areas over a longer period of time, but a larger number of samples would have to be tested to better understand the implications of MLVA in such a setting.
Footnotes
Authors' Contributions
U.G.B. performed a major portion of the laboratory work and analysis and interpretation of data and drafted the manuscript. R.K. contributed to the laboratory work and performed the analysis and interpretation of MLVA. M.K. formulated the main research hypotheses, supervised the project and fieldwork, and drafted the manuscript. N.K. conducted the fieldwork (including collection of animal and tick samples) and performed the laboratory analysis and analysis of patient data. T.C. performed cultivation and whole-genome sequencing analysis and interpretation of data. T.K. conducted the fieldwork (including collection of animal and tick samples) and laboratory analysis. M.P. conceptualized the work. T.A.-Ž. advised the project team and provided expertise on the laboratory work. All authors have read, revised, and approved the final version of the manuscript.
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This work was supported by the Slovenian Research Agency (grant no. P3-0083).