
Abstract
Coxiella burnetii is an obligate intracellular pathogen and the causative agent of Q fever. In Ukraine, 28 human cases of Q fever were reported between 1997 and 2006; however, there are no state-approved, standardized molecular diagnostic assays that can be used systematically to investigate C. burnetii transmission to humans and its distribution throughout Ukraine. To address this deficiency, we followed the recommendation of the World Organization for Animal Health (OIE) and developed a confirmatory PCR for C. burnetii for veterinary diagnosis in Ukraine. The PCR assay targeted the outer membrane-associated gene com1 in C. burnetii. Oligonucleotide primers were selected that amplify a 689-bp DNA fragment of the com1 gene (primers: CoxF2 = 5′-ACYGCAGGCGTGGCGATAG-3′ and CoxR4 = 5′-TGAAGGTTTTGTTGTGAGGTGGC-3′). The assay proved highly sensitive and specific to C. burnetii DNA detection (LOD = 0.37 pg/μL). Reproducibility of the test was verified by comparing the PCR results with those of a different PCR protocol and qPCR. Using the CoxF2/CoxR4 primer set and reaction conditions described here, the PCR Diagnostic Kit C. burnetii-PCR-TEST was developed and officially registered for use in Ukraine by the State Scientific Control Institute of Biotechnology and Strains (Kyiv, Ukraine) for diagnostic purposes.
Introduction
C oxiella burnetii is an obligate intracellular bacterium that causes Q fever in animals and humans (Karysheva 2002, Arricau-Bouvery et al. 2006, Bessarabov et al. 2007). Since 2007, the Data Analysis Office of the European Food Safety Authority (EFSA) has documented a significant spread of Q fever in humans in the European Union. The EFSA reports show a clear upward trend between 2007 and 2010 (2007: 585 cases; 2008: 1594 cases; 2009: 2719 cases; and 2010: 1380 cases), before declining between 2011 (759 cases) and 2012 (692 cases), and subsequently ramping up again between 2013 and 2016 (2013: 716 cases; 2014: 780 cases; 2015: 822 cases; and 2016: 1057 cases) (European Food Safety Authority 2010a, 2010b, European Food Safety Authority and European Centre for Disease Prevention and Control 2014, 2015, 2016, 2017). The Q-fever epizootic situation in countries bordering Ukraine (i.e., Poland, Slovakia, Hungary, and Romania) has been suggested to represent a significant risk of importation of the pathogen into the Ukrainian territory (Karysheva 2002, Bessarabov et al. 2007, European Food Safety Authority 2010a, 2010b). In Ukraine, data on Q fever in humans are published sporadically. In particular, 28 human cases of Q fever have been reported for the period between 1997 and 2006 at the national level. From 2007 to 2012, 10 cases of Q fever have been confirmed in eastern Ukraine in Donetsk Oblast (Kushnir et al. 2015). In 2011, 15 cases of Q fever were registered in Ukraine (Vynograd and Skal'ska 2013), and these values decreased to 4 cases in 2012 and 1 case in 2013.
The period of 2013–2016 with no reported new cases was considered as an interepidemic period. In subsequent years, however, an epidemic was observed in the western oblasts of Ivano-Frankivsk, Ternopil, Zakarpattia, and Lviv, as well as in central Ukraine in Cherkasy and Poltava oblasts (Kushnir et al. 2015, Zarichna 2018).
The spread of Q fever was further investigated in Ukraine using C. burnetii immunodetection. This assessment in humans identified positive serological samples in 15 to settlement of 11 rayons of Kyiv Oblast (north-central region) in 2009 (Komarenko 2010). Serology results identified nine enzootic areas for Q fever in the western part of the country, within six administrative rayons of Ternopil Oblast. The test population included 154 subjects, with 55.4% of them representing at-risk groups (i.e., those whose residence or occupation was associated with potential exposure to tick vectors carrying C. burnetii) (Kushnir et al. 2015). In the south, different rayons in Odessa Oblast accounted for 16 Q fever human cases between 2013 and 2014 (i.e., Danube–Dniester interfluve, Artsyz, Tarutyne, Tatarbunary, and Kiliya rayons). Seven individuals in these rayons developed the acute form of the disease (Beck et al. 2015).
Assessment of vector infectivity revealed the persistence of C. burnetii in Ixodes spp. ticks in Donetsk Oblast in eastern Ukraine (Kushnir et al. 2015). C. burnetii DNA was also detected in 2.82 ± 0.1% of ticks collected from domestic animals (dogs and cats) in Kyiv Oblast. The presence of C. burnetii DNA in ticks indicates establishment of the anthropogenic zoonotic foci in the area (Komarenko 2010, Vynograd and Komarenko 2013). Within a larger time frame (2010–2014), C. burnetii DNA was detected in four types of ticks in the Odessa Oblast: Ixodes ricinus (26.9 ± 8.7%), Hyalomma plumbeum (7.7 ± 5.2%), Dermacentor marginatus (61.5 ± 9.5%), and Dermacentor reticulatus (7.7 ± 5.2%). Detection of C. burnetii DNA was not restricted to ticks. The DNA was also detected in small rodents (Zarichna et al. 2015).
Livestock are susceptible hosts of C. burnetii infection. Between 2008 and 2014, monitoring of livestock by the State Scientific Research Institute of Laboratory Diagnostics and Veterinary and Sanitary Expertise (SSRILDVSE) showed a 24.62% seropositivity in the tested population. The geographic distribution of infected livestock indicated the presence of a natural foci in five rayons of Odessa Oblast (Artsyz: 26.9%, Kiliya: 4.9%, Tatarbunary: 4.6%, Bolhrad: 4.5%, and Tarutyne: 7.6%) (Marushchak 2016). An observed decline in human Q fever cases in Ukraine and in other countries may reflect the limited availability of serological and molecular diagnostic tests for Coxiella (Beck et al. 2015), rather than an actual decline in infection rate.
Since Ukraine lacks a state-approved, standardized molecular diagnostic assay for C. burnetii, direct comparison of diagnostic data from different regions of Ukraine may not be possible due to differences in assay sensitivity and specificity. The OIE recommends using PCR as a confirmatory detection method for acute infections. The lack of diagnostic tests and the growing need to monitor C. burnetii spread in the Ukraine motivated us to develop a sensitive and reliable diagnostic kit for C. burnetii for veterinary use nationwide. The optimized test that we developed, namely Coxiella burnetii-PCR-TEST, is described here and has been officially registered in the State Scientific Control Institute of Biotechnology and Strains in Kyiv, Ukraine, under the registration certificate number BB-00733-06-14 of 07/17/2015. This kit can now be used at the national level and has the potential to provide reliable PCR data to study the epidemiology of C. burnetii throughout the Ukraine.
Materials and Methods
Primer design
Recommended genes for the detection of C. burnetii are as follows: the superoxide dismutase (Sod B) gene, the heat shock operon encoding two heat shock proteins (htpA and htpB), and macrophage infectivity potentiator protein (cbmip); the isocitrate dehydrogenase gene (icd), an outer membrane protein coding gene (com1), and an insertion element (IS1111) were investigated for choice of target gene (Bruin de et al. 2009, Schimmer et al. 2012, OIE 2018). The com1 gene was chosen as target gene for developing primers for the C. burnetii PCR. We conducted sequence alignment comparing nucleotide sequences from the highly conserved single copy outer membrane-associated com1 protein of C. burnetii (GenBank Accession numbers: AB004693; AB004694; AB004695; AB004696; AB004697; AB004698; AB004699; AB004700; AB004701; AB004702; AB004703; AB004704; AB004705; AB004706; AB004707; AB004708; AB004709; AB004710; AB004711; AB004712; AF317646; AF317647; AF318145; AF318146; AF318147; AF318148; AF318149; HM237793; JX131361; JX131362; JX131363; JX131364; JX131365; JX131366) using the Vector NTI Advanced software, v. 10 (Invitrogen) (Marushchak 2013, Marushchak et al. 2014). Regions of 100% homology were used as target sequences for each potential assay. Phylogenetic sequence alignment of com 1 for the selected strains (codes listed above) was carried out using Geneious software, v.1.6.0_35-b 10 (Biomatters Ltd., New Zealand). Genbank homology search was performed against the selected PCR target sequence in com1 to rule out any possibility of unspecific hybridization of the target sequence with the host or other unrelated genomic sequences. The selected PCR primer set consists of the forward primer CoxF2 (5′-ACYGCAGGCGTGGCGATAG-3′) and the reverse primer CoxR4 (5′-TGAAGGTTTTGTTGTGAGGTGGC-3′) that generate a 689-bp amplicon.
Controls
Two positive controls were used to validate the PCR protocol: (i) DNA from C. burnetii strain 5131 (Genekam Biotechnology AG, Germany) and (ii) DNA from C. burnetii strain Gritta (Q-fever serological kit reagent, All-Russian Research Institute for Animal Health, Russian Federation). UltraPure DNase/RNase-Free Distilled Water (Invitrogen) or Nuclease Free Water (Qiagen, Germany) was used as negative control. DNA isolation was performed using High Pure PCR Template Preparation Kit (Roche, Germany).
Optimization of PCR conditions
All primers were synthesized commercially (Integrated DNA Technologies). The optimization of PCR amplification was accomplished by adjusting Mg2+ and dNTP concentrations in the PCR mixture and testing different annealing temperatures. We performed an annealing gradient experiment for primers CoxF2/CoxR4 at temperatures of 60°C, 62°C, 64°C, and 65°C. The PCR products were resolved by 1.5% agarose gel electrophoresis, stained with ethidium bromide, and visualized under UV light using the Gel Doc XR + Gel Documentation System (Bio-Rad). We also evaluated different concentrations of MgCl2 (i.e., 15, 10, and 5 mM). Amplification was performed using the PCR master mix by “AmpliSens-200-1” (Russian Federation, Moscow). The PCR master mix consists of two PCR mixes as follows: PCR mix 1 (lower phase) and PCR mix 2 (upper phase). PCR mix 1 containing volume of reagents for one reaction: 1 μL of 1.76 mM dNTPs, by 0.25 μL–100 pmol/μL of forward primer and reverse primer, and 3.5 μL of Nuclease Free Water. PCR mix 2 containing: 5 μL of 15 mM Mg2+, 0.5 μL–5 U/μL of DiaTaq polymerase, and 11.5 μL of Nuclease Free Water. The PCR reagents and enzyme were combined to 3 μL of test DNA (or DNA control) in a 25 μL final reaction volume. Thermocycling conditions were: 95°C for 4 min followed by 35 amplification cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s, followed by final elongation step at 72°C for 4 min and holding temperature of 4°C at the end of the run. Conventional PCRs were conducted using thermocycler Mastercycler (Eppendorf AG, Germany). Size verification of all PCR products in the study was performed using GeneRuler 100 bp Plus DNA Ladder (Thermo Fisher Scientific).
Specificity and sensitivity of primers
Specificity of the designed primers was evaluated using a panel of DNA samples of various pathogens that were regarded as potentially present in the samples that would be tested. The specificity was evaluated using reference control DNA C. burnetii#5131 and C. burnetii Gritta strains. Nontarget organism panel included the following: Salmonella typhimurium, Leptospira grippotyphosa, Brucella abortus, Chlamydophila psittaci, Listeria monocytogenes, Bovine herpesvirus 1, Pasteurella multocida, Escherichia coli, and Staphylococcus aureus.
Sensitivity of the primers was investigated using a panel of DNA samples, which contained different amounts of C. burnetii DNA. We prepared serial 10-fold dilutions of total DNA extracted from the C. burnetii Gritta strain. The DNA concentrations tested ranged from 37 ng/μL to 0.37 pg/μL. Concentration and purity of all DNA were evaluated using the spectrophotometer/fluorometer BioPhotometer 6131 (Eppendorf AG).
Independent validation of the C. burnetii PCR test by interlaboratory comparison tests
Independent validation of the test was performed by comparing PCR results using our primer set (i.e., CoxF2/CoxR4) with two independent PCR primer sets previously used for C. burnetii detection. The additional primer tests used in the comparison were as follows: Comtrg_f/Comtrg_r primer set—forward primer comtrg_f = 5′-CCCTGCAATTGGAACGAAG-3′ and reverse primer comtrg_r = 5′-GTTCTGATAATTGGCCGTCGACA-3′. This com1 gene primer set yields a 775-bp amplicon. This primer set was developed by the National Institute for Public Health and the Environment (RIVM), Centre for Infectious Disease Control, Laboratory for Zoonoses and Environmental Microbiology in the Netherlands (Bruin de et al. 2011). qPCR kit VetMax Screening Pack—Ruminant Abortion (Thermo Fisher Scientific).
A panel of C. burnetii DNA samples used in the comparison experiments was provided by the National Veterinary Research Institute (NVRI, Pulawy, Poland) in 2014 and 2015 as a part of an interlaboratory comparison test effort. Work was completed at SSRILDVSE, Research Department of Molecular and Genetic Studies.
Results
Primer design
We collected 34 GenBank nucleotide sequence entries for the highly-conserved, single-copy, outer membrane-associated com1 gene. This gene encodes a 27-kDa C. burnetii protein. Alignment of these multiple DNA sequences with the Geneious software identified a conserved com1 target sequence that was used for the selection of our PCR primer set sequence (CoxF2/CoxR4).
A phylogenetic tree was constructed using a Neighbor-joining tree build method. This genomic analysis of C. burnetii strains and isolates concluded that the region selected to generate the diagnostic primer isolate AB004712 contains a conserved com1 sequence present in all isolates and strains of the C. burnetii sequences evaluated (Fig. 1). For primer design, we used a region of the C. burnetii com1 gene having 99–100% of DNA sequence homology.
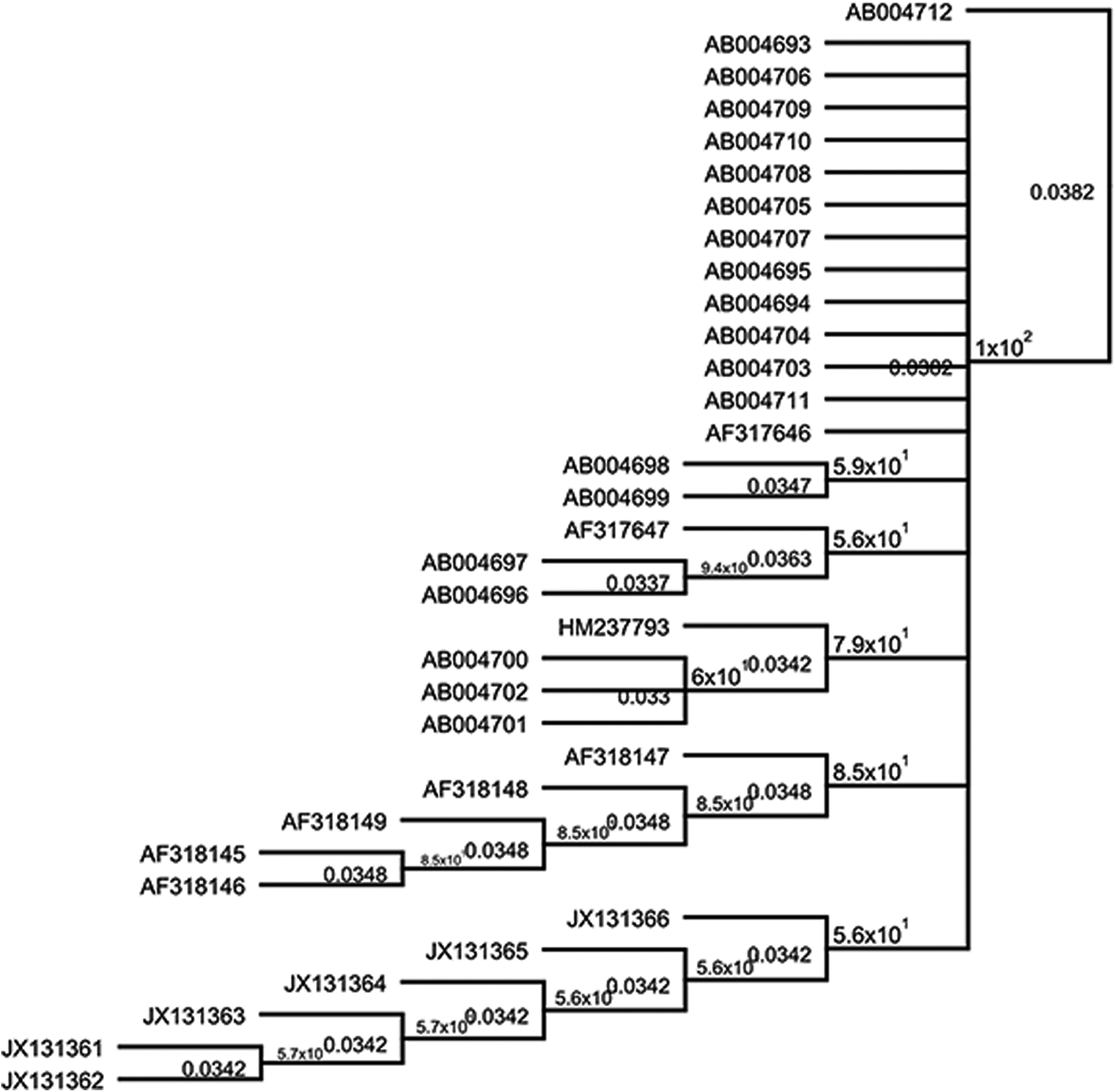
Neighbor-joining phylogenetic tree of Coxiella burnetii com1 gene. The tree was generated using the Geneious software based on the Tamura-Nei model genetic distance matrix values. The AB004712 was used as an outgroup. The numbers at the nodes indicate the percentage of 1000 bootstrap replicates that supported the interior branches.
According to the results of the phylogenetic analysis, the PCR primers for CoxF2 and CoxR4 were selected using the 5′-end of the com1 gene. The forward primer CoxF2 (5′-ACYGCAGGCGTGGCGATAG-3′) has a primer melting temperature (T m) of 58.4°C and a GC content of 65.8%; length of sequences is 19. The reverse primer CoxR4 (5′-TGAAGGTTTTGTTGTGAGGTGGC-3′) has a T m of 57.7°C and a content of 48.7%; length of sequences is 23. The CoxF2 primer contains a degenerate Y (pYrimidine) nucleotide corresponding to cytosine or thymine. This degenerate nucleotide in the com1 primer allows for the different genotypes of the pathogen to be accounted for. The CoxF2 and CoxR4 primers span conserved nucleotides at position 316–334 bp and 982–1004 bp of the com1 gene, respectively.
Amplification of an expected 689-bp DNA fragment for this PCR was confirmed by agarose gel electrophoresis using C. burnetii #5131 DNA.
Optimization
To determine the optimal primer concentrations for use in the PCR, each primer was tested using concentrations ranging from 10 to 25 pmol/μL. Primer concentrations of 15 pmol/μL showed the best amplicon DNA product using both C. burnetii #5131 and C. burnetii Gritta strain DNAs and the AmpliSens-200-1 PCR master mix.
The next stage in developing our PCR diagnostic test included optimization of PCR amplification parameters (i.e., Mg2+ and dNTP concentrations and annealing temperature).
Amplification results showed that the best amplicon yield was produced at 15 mM Mg2+ concentration. From the annealing temperature tested (i.e., 60°C, 62°C, 64°C, or 65°C), 60°C or 62°C produced higher yields of the amplicon. In higher temperature settings (i.e., 64°C or 65°C), a decrease in product specificity was observed. The best results were found using the 60°C annealing temperature for the primer pair CoxF2/CoxR4 (Marushchak et al. 2014).
The optimized PCR cycling conditions for a reproducible amplification using CoxF2/CoxR4 primers were: 95°C, 4 min; 35 cycles at 94°C—30 s, 60°C—30 s, 72°C—30 s; followed by a final extension step at 72°C—4 min; and a holding temperature of 4°C.
Evaluation of PCR sensitivity and specificity for the diagnostic test kits
PCR specificity was evaluated for the designed primers using a panel of DNA samples of various pathogens (data not shown). The established method showed no cross-reactivity with genomic DNAs of eight pathogens (i.e., bacteria DNAs tested: L. grippotyphosa, L. monocytogenes, B. abortus, C. psittaci, S. typhimurium, E. coli, P. multocida, S. aureus; viral DNA: B. herpesvirus 1). All DNA samples were tested in triplicates.
Specificity of our PCR method was demonstrated using heterogenous DNA samples with the CoxF2/CoxR4 primer pair. When these DNA preparations were used, the characteristic 689-bp com1 DNA fragment was not amplified.
Reproducibility of the assay was also assessed using the CoxF2/CoxR4 primer set to amplify the target DNA. Fluorescence intensity and length of PCR products were compared using two positive control DNA samples (C. burnetii#5131 and C. burnetii Gritta), unspecific genomic DNA samples (i.e., L. grippotyphosa, L. monocytogenes, B. abortus, C. psittaci, S. typhimurium, E. coli, P. multocida, S. aureus, and B. herpesvirus 1), and a negative control sample (Nuclease Free Water; Qiagen). Each sample was tested in triplicate.
PCR sensitivity was confirmed as shown in Fig. 2. Amplification of PCR products was evaluated comparing log serial dilutions of C. burnetii #5131 DNA (i.e., 37, 3.7, and 0.37 ng/μL and 37, 3.7, and 0.37 pg/μL—lanes 1 through 6). This method showed a sensitivity of 0.37 pg/μL (Fig. 2, lane 6).
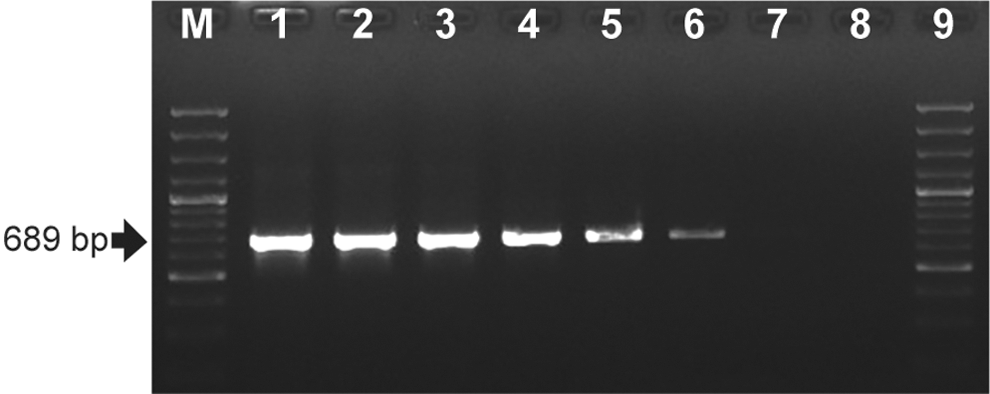
Sensitivity of the PCR detection method. Amplification of the 689-bp DNA com1 DNA fragment was assessed using log dilutions of the C. burnetii Gritta DNA. Lanes 1–6 show log serial dilutions of the DNA template (i.e., undiluted [37 ng/μL], 10−1, 10−2, 10−3, 10−4, and 10−5). Lanes 7 and 8 are the negative controls (no DNA). M = 100 bp plus DNA Ladder.
Specific amplicons were detected in all tested DNA concentrations of the C. burnetii Gritta DNA in Fig. 2, indicating the accuracy of the method. The test of sensitivity was repeated five times using different thermocyclers. The qualitative results were identical demonstrating the reproducibility of the test.
Thus, the results indicate high sensitivity and specificity of the designed CoxF2/CoxR4 primer set and the possibility of their use as diagnostic test kits.
Validation of the C. burnetii DNA detection method using primers CoxF2 and CoxR4
Validation of the C. burnetii DNA detection method using primers CoxF2 and CoxR4 was conducted at the Research Department of Molecular and Genetic Studies, SSRILDVSE, during intralaboratory comparison of the Q-fever test in 2014 and 2015.
We received an encrypted panel of six DNA samples from the National Reference Laboratory for Q fever (NVRI) to assay by our PCR method for the presence of the causative agent of Q fever. The study was conducted comparing amplifications using the designed primer pair CoxF2/CoxR4 with those using the primers comtrg_f/comtrg_r and those included in the qPCR kit VetMAX Screening Pack—Ruminant Abortion. C. burnetii #5131 DNA was used as positive control.
National Reference Laboratory for Q fever (NVRI) confirmed that the results were correct.
The observed 100% match indicates high sensitivity and specificity of the designed primers and the possibility of their use in the development of diagnostic test kits.
Discussion
Q fever is a zoonotic problem in many countries causing significant economic losses because infected farm animals may be less productive or die. Furthermore, there is an associated risk of human infection through various routes (e.g., airborne, oral, and vector borne), with people engaged in animal farming being particularly vulnerable (Babudieri 1959, Karysheva 2002, Kalinina 2004, Howe et al. 2009, Bruin de et al. 2011, Loginiv et al. 2015).
Natural Q fever foci have been identified in 10 oblasts of Ukraine, with active transmission over 40 years in the Autonomous Republic of Crimea, Dnipropetrovsk, and Lviv oblasts (Babudieri 1959, Kalinin 2004, Vynograd and Skal'ska 2013). In 2013–2014, Ivano-Frankivsk, Ternopil, Zakarpattia, Lviv, Cherkasy, and Poltava oblasts were reservoirs for C. burnetii between the epidemic seasons (Marushchak 2016). Odessa Oblast has an ongoing active Q fever epidemic (Beck et al. 2015). C. burnetii continues to exist in Donetsk Oblast in populations of Ixodid ticks (Kushnir et al. 2015). The epidemiological study by Kushnir et al. (2015) showed that the disease is associated with pets and farm animals and milk products and that the vector-borne mode of infection is also possible.
The Ukrainian Center of Rickettsial Infections (Lviv Institute of Epidemiology and Hygiene) identified 257 enzootic Q fever areas in 18 oblasts of the country, Crimea and Sevastopol (Zarichna 2018). The expected growth of animal farming and environmental management in recent years and an increasing number of landscaped gardens with their associated fauna (potential reservoir hosts and carriers of C. burnetii) contribute to the preservation of natural Q fever foci.
Social and economic conditions in Ukraine lay the foundation for the spread of Q fever among the human population. The lack of correlation between the relatively high level of detection of seropositive results in diagnostic tests and the low level of officially registered cases of Q fever is attributed to the limited number of research subjects and lack of diagnostic kits, which are common in Ukraine and some European countries. Currently, in Ukraine, Q fever diagnosis in animals is determined mainly by serological tests (immunosorbent assay [enzyme linked immunosorbent assay, ELISA] and complement fixation test [CFT]). These methods of diagnosis based on epidemiological data, isolation, and identification of the pathogen using bioprobe, ELISA, CFT, or indirect immunofluorescence assay (IFA) are time consuming and labor intensive and have certain disadvantages; for example, C. burnetii immunological detection methods using IFA and ELISA are insufficiently sensitive and specific and do not meet the requirements of the practical veterinary medicine (reference here or explain what you mean by requirement that do not meet practical vet med). These shortcomings prompted us to develop and introduce PCR-based test kits, which are characterized by high sensitivity and specificity and can be used as confirmatory tests for the presence of the pathogen.
Because there were no approved Ukrainian kits for diagnosis of the Q fever agent by PCR, it was necessary to import relatively expensive kits from other countries. The development of Ukrainian test kits will reduce cost to laboratory facilities conducting the tests and improve the quality of C. burnetii molecular diagnostics. The optimized test kit Coxiella burnetii-PCR-TEST was validated at the interlaboratory level for sensitivity and specificity compared with the qPCR kit VetMAX Screening Pack—Ruminant Abortion and a PCR protocol using the comtrg_f/comtrg_r primer set.
Conclusions
Bioinformatic analysis of the com1 gene sequence encoding the highly conserved, 27-kDA outer membrane protein of C. burnetii was conducted. In accordance with the test results, one primer pair design, CoxF2/CoxR4, produced a PCR amplification product 689 bp in length. Primer CoxF2 contains the degenerate nucleotide Y corresponding to cytosine or thymine, which allows for amplification of C. burnetii com1 gene sequences of different pathogen genotypes. PCR amplification parameters for C. burnetii DNA detection using the CoxF2/CoxR4 primer pair were developed, and PCR conditions were tested. Based on the results of the validation of the C. burnetii DNA detection method using the primer pair CoxF2/CoxR4, the document package for diagnostic kit Coxiella burnetii-PCR-TEST registration was developed and submitted. The diagnostic kit Coxiella burnetii-PCR-TEST has high sensitivity and specificity (patent # U201502187), serving as a valid diagnostic tool for C. burnetii detection in biological samples. The limit of detection determined by agarose gel electrophoresis is ≥0.37 pg/μL of C. burnetii DNA in the sample. The results of interlaboratory testing were confirmed by the National Reference Laboratory for Q fever (NVRI).
Footnotes
Acknowledgments
The authors acknowledge the director of SSRILDVSE for supporting this research. The authors acknowledge the United States Department of Defense, Defense Threat Reduction Agency (DTRA), and Cooperative Biological Engagement Program (CBEP) for their assistance and financial support in the publication of this article. While DTRA/CBEP did not support the research described in this publication, the Program supported the article publication. The contents of this publication are the responsibility of the author and do not necessarily reflect the views of DTRA or the United States Government. The authors thank Dr. Christopher Mores, Dr. Rebecca Christofferson, Dan Chisenhall, and Dr. Michael McCracken from Louisiana State University, School of Veterinary Medicine, Vector-Borne Disease Laboratories, United States of America for support in identifying the primer sequences and for providing the use of the Geneious software. The author is thankful to Dr. Stephen Higgs (Kansas State University, United States of America), Dr. Wojciech Iwaniak (Scientific Consulting, Pulawy, Poland), and Dr. Falgunee K. Parekh (Metabiota, Inc., United States of America) for their writing mentorship with this article.
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This research was conducted within the state funded scientific project of the State Scientific Research Institute of Laboratory Diagnostics and Veterinary and Sanitary Expertise “Development, study and comparison of different methods and tools for diagnosis, treatment and prevention of infectious and parasitic diseases of animals” 2009–2018 years number 0109U001081. Work was completed in 2009–2016 by the State Scientific Research Institute of Laboratory Diagnostics and Veterinary and Sanitary Expertise.