
Abstract
Bartonella spp. are parasites of mammalian erythrocytes and endothelial cells, transmitted by blood-feeding arthropod ectoparasites. Different species of rodents may constitute the main hosts of Bartonella, including several zoonotic species of Bartonella. The aim of this study was to identify and compare Bartonella species and genotypes isolated from rodent hosts from the South Sinai, Egypt. Prevalence of Bartonella infection was assessed in rodents (837 Acomys dimidiatus, 73 Acomys russatus, 111 Dipodillus dasyurus, and 65 Sekeetamys calurus) trapped in 2000, 2004, 2008, and 2012 in four dry montane wadis around St. Katherine town in the Sinai Mountains. Total DNA was extracted from blood samples, and PCR amplification and sequencing of the Bartonella-specific 860-bp gene fragment of rpoB and the 810-bp gene fragment of gltA were used for molecular and phylogenetic analyses. The overall prevalence of Bartonella in rodents was 7.2%. Prevalence differed between host species, being 30.6%, 10.8%, 9.6%, and 3.6% in D. dasyurus, S. calurus, A. russatus, and A. dimidiatus, respectively. The phylogenetic analyses of six samples of Bartonella (five from D. dasyurus and one from S. calurus) based on a fragment of the rpoB gene, revealed the existence of two distinct genetic groups (with 95–96% reciprocal sequence identity), clustering with several unidentified isolates obtained earlier from the same rodent species, and distant from species that have already been described (90–92% of sequence identity to the closest match from the GenBank reference database). Thus, molecular and phylogenetic analyses led to the description of two species: Candidatus Bartonella fadhilae n. sp. and Candidatus Bartonella sanaae n. sp. The identification of their vectors and the medical significance of these species need further investigation.
Introduction
B
In almost all countries, wild rodents are infected by an extremely diverse range of Bartonella species/strains and generally with a high prevalence. Examples include rodents in North America (Kosoy et al. 1997), Asia (Ying et al. 2002, Inoue et al. 2008, 2010), Australia (Gundi et al. 2009), Africa (Bajer et al. 2006, Sato et al. 2013), and Europe (Birtles et al. 1995, Tea et al. 2004, Welc-Faleciak et al. 2010, Paziewska et al. 2011). Prevalence of Bartonella infections in rodents ranges from 6% in the Greater Jakarta area in Indonesia (Winoto et al. 2005) to 70% in the Russian Far East (Mediannikov et al. 2005, Morick et al. 2011). To date, the arthropods that have been confirmed as competent vectors for Bartonella are the sand fly Lutzomyia verrucarum as a vector for Bartonella bacilliformis (Noguchi and Battistini 1926) and the louse Pediculus humanus humanus as a vector for B. quintana (Swift 1920). Moreover, recent studies have revealed that a range of flea species may also play a role as vectors of Bartonella: the flea Ctenophthalmus nobilis as a vector for B. grahamii and Bartonella taylorii (Bown et al. 2004), Ctenocephalides felis as a vector for B. henselae (Chomel et al. 1996), the fleas Megabothris turbidus, M. walkeri, Hystrichopsylla talpae, Ctenophthalmus assimilis, and Ctenophthalmus agyrtes as vectors for B. taylorii (Jusko 2015), and the fleas M. turbidus, M. walkeri and C. assimilis, as vectors of B. grahamii (Jusko 2015).
Because of the difficulties in identifying Bartonella and distinguishing between the species through the use of conventional microscopy and biochemical tests, molecular approaches have been developed based on PCR, followed by sequencing of several housekeeping genes, such as citrate synthase (gltA) and RNA polymerase beta subunit (rpoB) genes (Norman et al. 1995, Renesto et al. 2001, La Scola et al. 2003, Inoue et al. 2008). In particular, the molecular marker genes, rpoB and gltA, have been widely used for differentiating between species because of the much lower degrees of taxonomically relevant variability in these genes within Bartonella species (La Scola et al. 2003). Therefore, variation in the rpoB and gltA genes can be applied as a cutoff criterion for the designation of novel species in the genus Bartonella.
Our long-term studies on temporal changes in the hemoparasite community (Babesia, Bartonella, Haemobartonella, Hepatozoon, and Trypanosoma) of rodents in Egypt (Behnke et al. 2000, Bajer et al. 2006) have led to the description of a novel species—Babesia behnkei (Bajer et al. 2014a), and provided molecular evidence for the existence of two different variants of Hepatozoon and two novel variants of Trypanosoma acomys in Acomys dimidiatus (Alsarraf et al. 2016). The aim of the current study was to identify the Bartonella species/strains in rodent hosts, including Wagner's gerbil Dipodillus dasyurus and the bushy-tailed jird Sekeetamys calurus, inhabiting isolated montane valleys in the Sinai Massif, Egypt.
Materials and Methods
Field studies in Sinai, Egypt
Fieldwork was conducted over 4–5-week periods in August and September in 2000, 2004, 2008, and 2012 and was based at the Environmental Research Centre of Suez Canal University (2000, 2004) or at Fox Camp (2008, 2012) in the town of St. Katherine, South Sinai, Egypt. Trapping was carried out in four montane wadis (dry valleys) in the vicinity of St. Katherine. The local environment and general features of the four study sites (W. Arbaein, W. Gebal, W. Tlah, W. Gharaba), as well as their spatial relationships with one another, have been described previously (Behnke et al., 2000). Details of the number of rodents sampled by site and year of study are presented in Table 1. At each site, rodents were caught live in Sherman traps that had been placed among the rocks and boulders around walled gardens and occasionally along the lower slopes of wadis. These were set out at dusk and inspected in the early morning before exposure to direct sunlight. All traps were brought into the local or base camp, where the animals were removed, identified, and processed. Traps were reset the following evening.
At inspection, each of the four rodent species (A. dimidiatus, Acomys russatus, D. dasyurus, and S. calurus) was sexed, weighed, measured, and scrutinized for obvious lesions as described by Behnke et al. (2004). Ectoparasites visible during field examination were removed and placed into 70% ethanol. Blood and fecal samples were taken and animals were then either fur marked individually, or ear clipped and released close to the point of capture, or returned to the main camp at St. Katherine for autopsy. A maximum of 40% of the captured rodents from each site were culled (by agreement with the St. Katherine National Protectorate authorities).
Blood collection and DNA extraction
Thin blood smears were prepared from drops of blood taken from the retro-orbital plexus using heparinized capillary tubes of animals lightly anesthetized with ether during examination in the field and from the heart of those that were autopsied under terminal anesthesia with ether. Blood smears were air-dried, fixed in absolute methanol, and stained for 1 h in Giemsa stain in buffer at pH 7.2 or by Hemacolor stain (Merck, Germany). Each smear was examined under oil immersion ( × 1000 magnification). Erythrocytes infected with Bartonella spp. were counted in 200 fields of vision. Microscopical observation of stained blood smears was used as the only detection method for study in 2000. Thereafter, from 2004 onward, molecular techniques were also used. For these, 200 μL of whole blood was collected into 0.001 M EDTA from all the culled animals and frozen at −20°C. Total DNA was extracted from whole blood using the DNeasy Blood and Tissue kit (Qiagen) and stored at a temperature of −20°C.
Molecular characterization
For the amplification of the rpoB gene fragment, we used the primers 1400F 5′-CGCATTGGCTTACTTCGTATG-3′ and 2300R 5′-GTAGACTGATTAGAACGCTG-3′ (Renesto et al. 2001). The PCR program was modified starting by denaturation for 5 min in 95°C, followed by 38 cycles (95°C for 45 s, 55°C for 45 s, and 72°C for 45 s), and then an extension step at 72°C for 7 min. For the gltA gene, we used the primers CS140f 5′-TTACTTATGATCC(GT)GG(CT)TTTA-3′ (Birtles and Raoult 1996) and CS1137r 5′-AATGCAAAAAGAACAGTAAACA-3′ (Norman et al. 1995). The PCR was started by denaturation for 2 min at 95°C, followed by 40 cycles of 1 min at 95°C, 1 min at 54°C, and 2 min at 72°C, with a final extension step of 7 min at 72°C. Reactions were performed in a 20 μL volume, containing 1 × PCR buffer, 0.2 U Taq polymerase (Thermo Scientific), 1 μM of each primer, 2 mM dNTP (A&A Biotechnology), and 2 μL of the extracted DNA sample. Negative controls were carried out in the absence of template DNA and the positive control was genomic DNA of B. grahamii from our earlier study (Alsarraf 2012, Bajer et al. 2014b). PCR products were subjected to electrophoresis on a 1.5% agarose gel, stained with Midori Green stain (Nippon Genetics GmbH), and sequenced by a private company (Genomed S.A., Poland).
Genotyping and phylogenetic analysis
Analysis of 860-bp rpoB gene fragment
From the eight Bartonella-positive rodent samples (Table 2), six Bartonella rpoB sequences were obtained from D. dasyurus and one from S. calurus (Table 2). In addition, one Bartonella sample obtained from A. dimidiatus from W. Tlah was genotyped by the amplification and sequencing of a shorter 333-bp fragment of the rpoB gene (Paziewska et al. 2011). All obtained sequences were aligned using MEGA v. 6.0 (Tamura et al. 2013). All our consensus sequences have been deposited in the GenBank database (accession numbers KX244342, KX244343, KX244344, KX244344, and KX244346). Akaike information criterion was used in the jModel Test to identify the most appropriate model of nucleotide substitution. A representative tree for rpoB sequences was constructed using MEGA v. 6.0, applying the maximum likelihood method and a Hasegawa–Kishino–Yano (I+G) model (Fig. 2).
Analysis of 810-bp gltA gene fragment
The analysis of eight Bartonella-positive rodent samples (Table 2) yielded four Bartonella gltA sequences from D. dasyurus and one sequence from S. calurus (Table 2). All obtained sequences were aligned using MEGA v. 6.0 (Tamura et al. 2013). All obtained sequences of Bartonella spp. have been deposited in the GenBank database (accession numbers KX137114, KX137115, KX137116, KX137117, and KX137118). Akaike information criterion was used in the jModel Test to identify the most appropriate model of nucleotide substitution. A representative tree for gltA sequences was obtained using the maximum likelihood method and a Hasegawa–Kishino–Yano (+G) model (Fig. 3).
Statistical analyses
Prevalence (percentage of animals infected) was calculated based on microscopical observations and PCR results. For analysis of prevalence, we used maximum likelihood techniques based on log-linear analysis of contingency tables in the software package IBM SPSS (version 21.0.; IBM Corp). Initially, a full factorial model was fitted, incorporating as factors host species (four levels), year of study (four levels, each of the four surveys), and site (four levels, the four wadis). The presence of Bartonella was implemented as “infection” and was considered as a binary factor (two levels, present or absent).
Ethical issue
Rodents from St. Katherine National Protectorate were sampled by agreement with the St. Katherine National Protectorate authorities obtained for each phase of the fieldwork. A maximum of 40% of the captured rodents from each site were culled by agreement with the St. Katherine National Protectorate authorities.
Results
Family Bartonellaceae, Gieszczykiewicz 1939 (Vos et al. 2009)
Genus Bartonella Strong et al. 1915 (Vos et al. 2009)
Candidatus Bartonella fadhilae n. sp.
Type-hosts: Wagner's gerbil, D. dasyurus (Rodentia, Muridae, Gerbillinae) and bushy-tailed jird, S. calurus (Rodentia, Muridae, Gerbillinae).
Type-locality: Wadi Gebal in the Sinai Mountains, Egypt.
Type-specimens: I.91 from D. dasyurus, sampled on August 21, 2012, in W. Gebal, Sinai Mountains, Egypt, and I.122 from S. calurus sampled on August 23, 2012, in W. Gebal, Sinai Mountains, Egypt.
Vector: Currently unknown.
Representative sequences: GenBank KX244344 and KX244346 (rpoB gene); KX137116 and KX137118 (gltA gene).
ZooBank registration: Candidatus Bartonella fadhilae n. sp. act:A9459D34-9962-4DA6-B82B-B748277602CF
Candidatus Bartonella sanaae n. sp.
Type-host: Wagner's gerbil, D. dasyurus (Rodentia, Muridae, Gerbillinae)
Type-locality: Wadi Gebal in the Sinai Mountains, Egypt.
Other localities: Wadi Arbaein in Sinai Mountains, Egypt.
Type-specimens: I.59 from D. dasyurus, sampled on August 21, 2004, in W. Gebal, Sinai Mountains, Egypt, and I.49 from D. dasyurus, sampled on August 18, 2012, in W. Arbaein, Sinai Mountains, Egypt.
Vector: Currently unknown.
Representative sequences: GenBank KX244343, KX244342 (rpoB gene), KX137114, KX137115 (gltA gene).
ZooBank registration: act:76DC10B7-9536-4600-930A-2920DD483C3F
Morphological characteristics
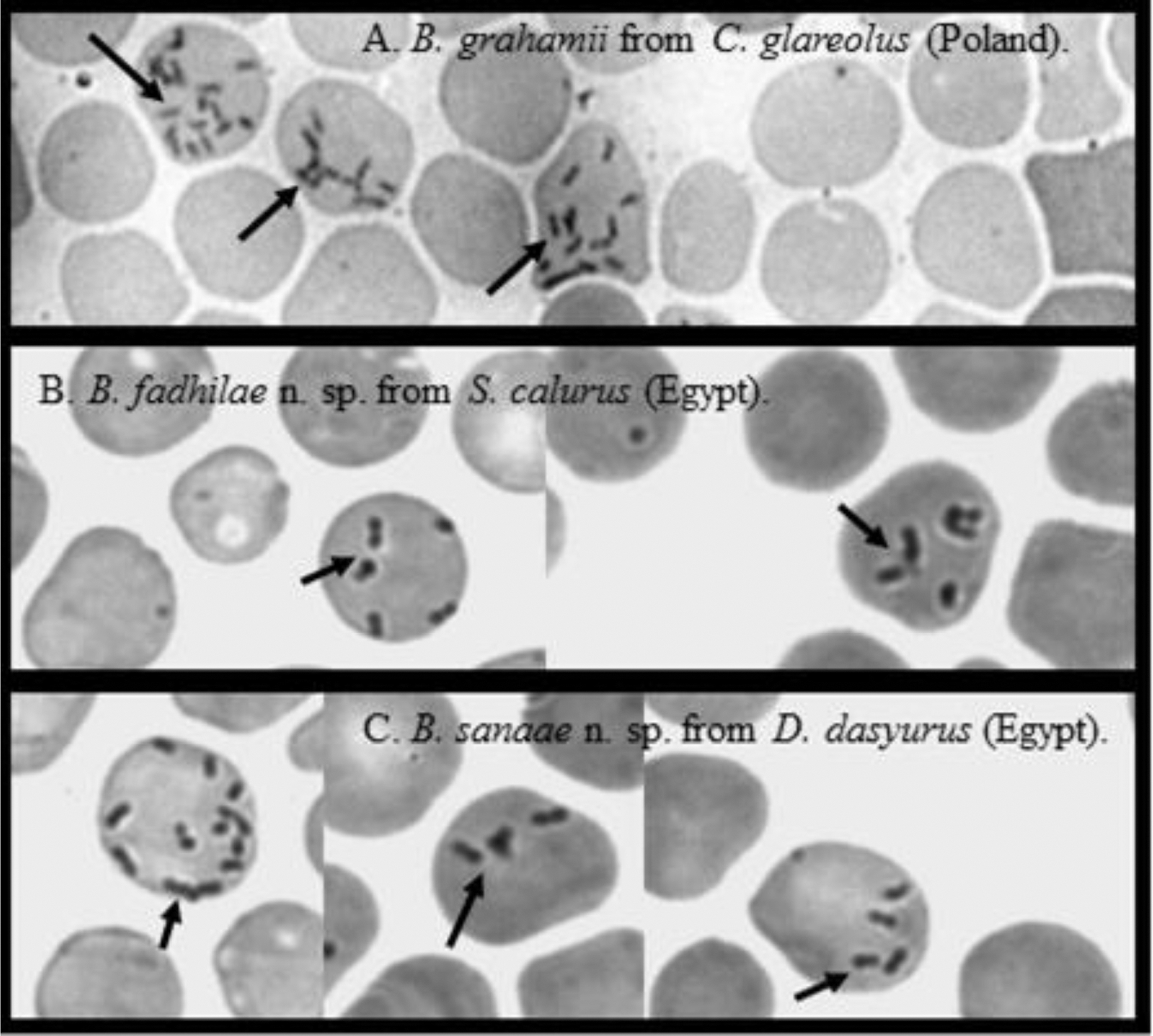
Microscopic examination of blood smears revealed that Candidatus Bartonella fadhilae n. sp., Candidatus Bartonella sanaae n. sp., and other samples of Bartonella sp. were indistinguishable morphologically as viewed through conventional microscopy (Fig. 1). Individual bacteria appeared as thin, short, pleomorphic bacilli or coccobacilli and were also morphologically indistinguishable from other Bartonella spp., known to occur in rodent hosts (Fig. 1).
Field studies: spatiotemporal dynamics of Bartonella infection in rodents
Altogether, 1086 rodents from the Sinai Mountains, Egypt, were sampled in four montane valleys (wadis) in 2000, 2004, 2008, and 2012, including 837 individuals of the spiny mouse A. dimidiatus, 111 Wagner's gerbils D. dasyurus, 73 A. russatus, and 65 bushy-tailed jirds S. calurus (Table 1). Overall prevalence of Bartonella spp. was highest in Wagner's gerbils (30.63%) in comparison with A. dimidiatus (3.58%) or A. russatus (9.58%) and S. calurus (10.76%) (host species × Bartonella infection: χ2 = 59.3, df = 3, p < 0.001) (Table 1). The highest prevalence of Bartonella infection in the rodent communities from the Sinai was recorded in 2004—14.33% (year of study × Bartonella infection: χ2 = 47.221, df = 3, p < 0.001). Infections with Bartonella spp. were identified in all host populations and in all study sites, but we noted the highest prevalence in W. Gebal in three surveys 2000, 2008, and 2012 and the lowest was in W. Gharaba in 2000, 2004, and 2008 (year of study × site × Bartonella infection: χ2 = 28.1, df = 9, p = 0.001) (Table 1).
Molecular identification of Bartonella sp. from A. dimidiatus, D. dasyurus, and S. calurus
We amplified the DNA of Bartonella spp. from eight rodent samples, six were from D. dasyurus, one from S. calurus, and one from A. dimidiatus (Table 2). Good quality sequences of both molecular markers rpoB and gltA were obtained only for three rodents: samples A and B from D. dasyurus and sample C from S. calurus (Table 2). Bartonella samples from the remaining rodents (D–H) were genotyped based only on a single marker (rpoB or gltA; Table 2).
Only one sample was successfully identified through a BLAST search in the GenBank database: sample F from A. dimidiatus was recognized based on the short fragment of the rpoB gene (333 bp). The BLAST revealed 99.37% identity of sample F to B. acomydis from A. russatus from Egypt (accession number AB602556) (Table 3).
Cov., coverage.
Among the seven remaining Bartonella samples, seven different variants with varying degrees of similarity were recognized based on two markers (Tables 3 and 4).
Sequences of the rpoB gene from samples A and D, both from Wagner's gerbil, showed 99.61% identity to each other and varied by only three nucleotides and, therefore, constituted a single group (R1) (Table 3 and Supplementary Fig. S1; Supplementary Data are available online at
The phylogenetic analyses of the rpoB sequences of our samples described in this work and the reference sequences from the GenBank database revealed the presence of three major groups (Fig. 2). In the first group our sequences clustered in two subgroups, R1 and R2, creating a polyphyletic group together with several unnamed isolates of Bartonella from S. calurus from Egypt (Inoue et al. 2009). The two subgroups of Bartonella sequences, R1 and R2, were clearly separated and distinct from each other, as confirmed by the significant bootstrap support (Fig. 2). Almost all of the Bartonella spp. in the phylogenetic tree that we derived are from rodents, and include two sequences from pathogenic species: B. elizabethae from humans from the United Kingdom and B. bacilliformis, but both are distinct from Bartonella spp. described by us.
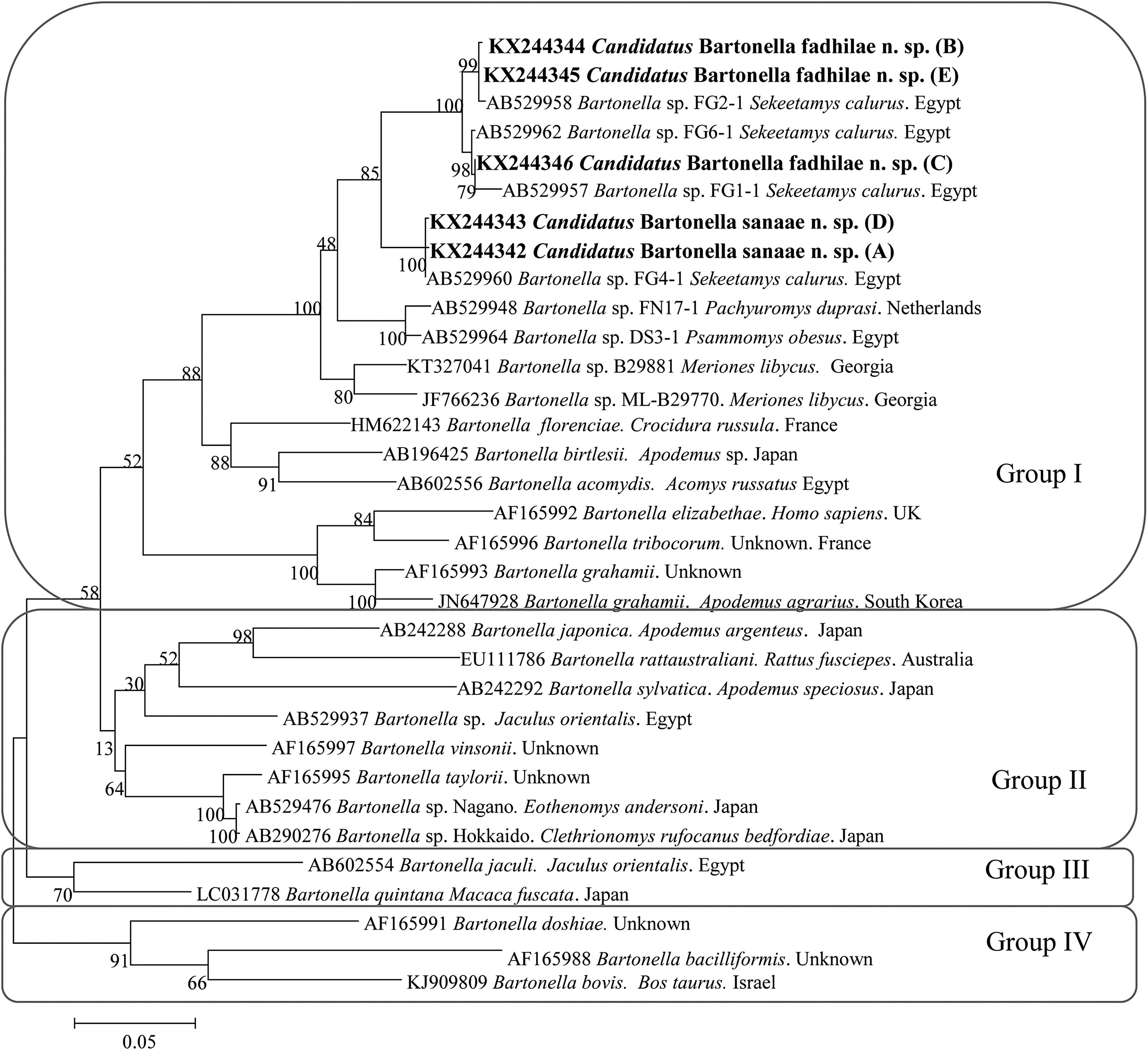
The phylogenetic tree of Bartonella spp., based on a fragment of the rpoB gene, was inferred using the maximum likelihood method and a Hasegawa–Kishino–Yano (G+I). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The analysis involved 33 nucleotide sequences. All positions containing gaps and missing data were eliminated. Evolutionary analyses were conducted in MEGA6.0.
As the analysis of the gltA sequences showed high similarity among sequences A, B, and G (98.7–99.5%) and our sequences differed only by 4–10 nucleotides (Table 4 and Supplementary Fig. S2), they constitute one group of Bartonella variants (G1). Interestingly, all these sequences were obtained from one host species, D. dasyurus from W. Gebal (Table 3). A BLAST search revealed that the G1 group showed the highest similarity (91–93%) to B. acomydis from A. russatus from Egypt, and our sequences differed by 18–23 nucleotides compared with the reference sequence of B. acomydis (AB602553) (Supplementary Fig. S2).
The gltA sequence of the sample C from S. calurus (Table 2) showed 95–96% identity to the sequences from the G1 group (26 nucleotide differences), and displayed also the highest identity (90.85%) with B. acomydis from A. russatus from Egypt (15 nucleotide differences in relation to the B. acomydis reference sequence AB602553) (Table 4 and Supplementary Fig. S2).
The gltA sequence of the unique sample H (from D. dasyurus from W. Gharaba) was quite distinct from all other gltA sequences, displaying a low similarity of 93–96% to our sequences, with 28–41 nucleotide differences, and 93% identity to B. acomydis from A. russatus from Egypt with 14 nucleotide differences (Table 4 and Supplementary Fig. S2).
The phylogenetic analyses of the gltA sequences revealed four main groups (Fig. 3). The samples belonging to the abovementioned group G1 clustered together with samples C and H, and created a polyphyletic subgroup, which is distinct from all other representatives within this group. Interestingly, all Bartonella spp. in this group are from rodents originating from different regions of the world (Fig. 3), while all other main groups contain a range of Bartonella species from different hosts and every group contains at least one pathogenic Bartonella, distinct from our sequences.
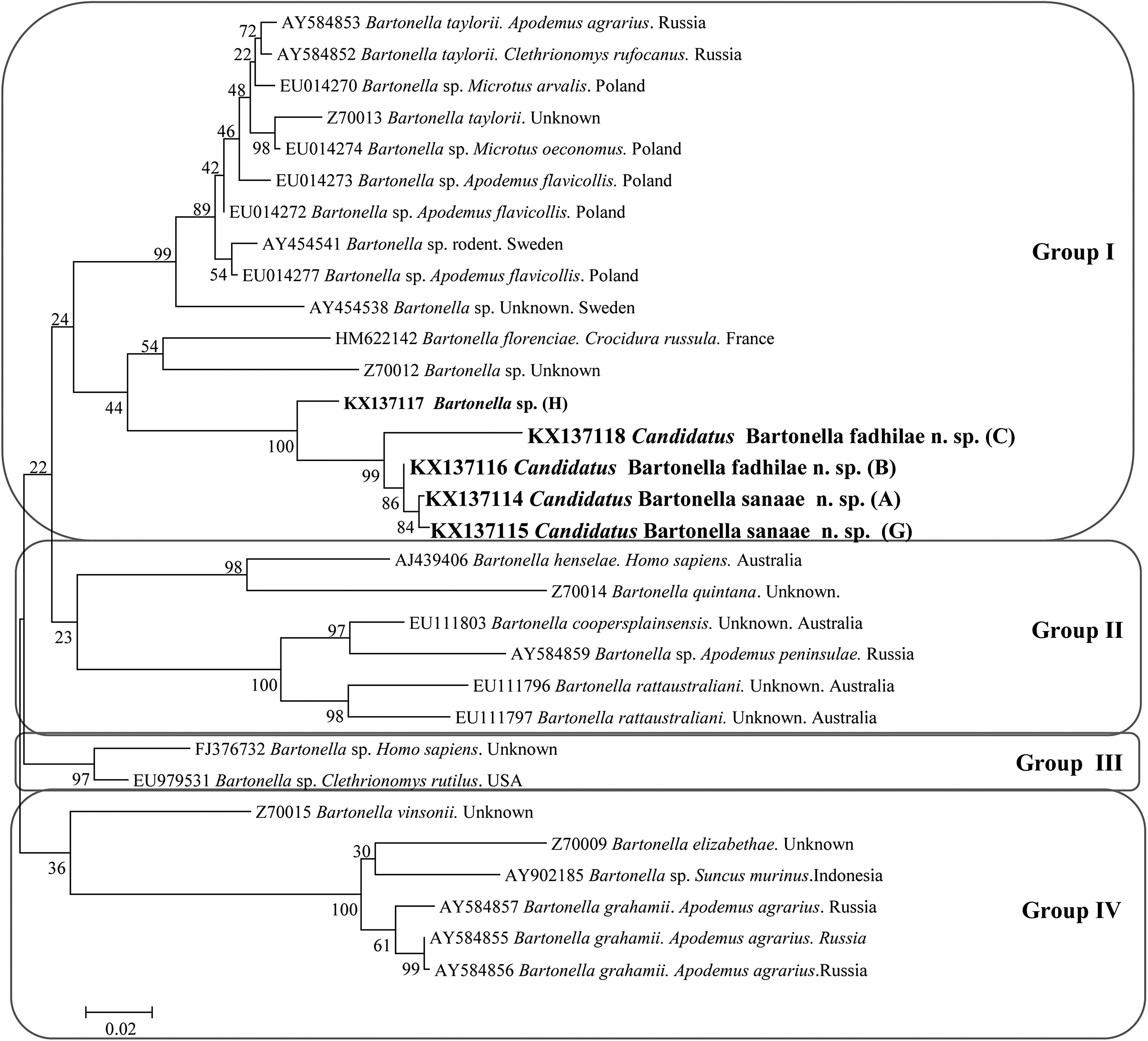
The phylogenetic tree of Bartonella spp., based on a fragment of the gltA gene, was inferred using the maximum likelihood method and a Hasegawa–Kishino–Yano (G). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The analysis involved 31 nucleotide sequences. All positions containing gaps and missing data were eliminated. Evolutionary analyses were conducted in MEGA6.0.
Discussion
Molecular and phylogenetic analyses of the Bartonella spp. infecting Wagner's gerbil and the bushy-tailed jird from the Sinai Mountains revealed their distant relationships to all known species in this genus, and therefore, two novel species of Bartonella are proposed. Infections with the Candidatus Bartonella fadhilae n. sp. were found in two host species D. dasyurus and S. calurus, and Candidatus Bartonella sanaae n. sp. was found only in D. dasyurus. Morphologically there were no differences between the two novel species of Bartonella.
Our study has two pertinent limitations, the principal being a lack of conventional microbiological description based on cultured isolates; none of the samples in our study was maintained as an axenic isolate. However, our samples were collected from rodents trapped in remote sites in the deserts of the southern Sinai, and there was no possibility for initiating bacterial cultures in that setting. However, almost all known Bartonella species share very similar biochemical activity, as in other groups of organisms, for this genus also new species descriptions are mainly based on molecular data (Birtles et al. 1995, Gundi et al. 2004, Inoue et al. 2010, Mediannikov et al. 2013, Sato et al. 2013). Second, while the study could have benefitted from an multilocus sequence analysis, we were only able to use two genetic markers because our attempts to amplify several other genetic markers using published sets of primers were unsuccessful (16S rDNA, ribC, ftsZ (Paziewska et al. 2011) and gro EL (Zeaiter et al. 2002). Nevertheless, our study clearly emphasizes the genetic diversification of Egyptian Bartonella from the species/strains of Bartonella described and recognized from other regions of the world.
The rpoB and gltA genetic markers enable a clear discrimination of the known Bartonella species (Gundi et al. 2009). Molecular analyses of the rpoB sequences from this and previous studies (Inoue et al. 2009) have revealed that there are two variants of Candidatus Bartonella fadhilae n. sp.: one infecting two host species D. dasyurus and S. calurus, and the second one that has been identified so far in only one host species D. dasyurus (Fig. 2). All our rpoB variants of Candidatus Bartonella fadhilae n. sp were obtained from rodents trapped in one valley, W. Gebal in 2004 and 2012, which may suggest that it is an endemic population of Bartonella spp. In the case of Candidatus Bartonella sanaae n. sp., we also found two rpoB variants, both from the same host species (D. dasyurus) but from two isolated montane valleys, W. Gebal (2004) and W. Arbaein (2012) (Fig. 3).
It has been reported previously that the Bartonella isolates should be considered to represent a novel species, if the hypervariable 327 bp gltA gene fragment shares <96% sequence similarity and if an 825 bp rpoB fragment shares <95.4% sequence similarity with sequences from strains of species with validly published names (La Scola et al., 2003, Gundi et al., 2009). In the current study, we amplified and analyzed a near full-length (810 bp) fragment of the gltA gene with identity below 91% to B. florenciae, and an 860 bp long fragment of the rpoB gene with identity below 92% to B. florenciae. To check, if these differences were spread throughout the whole gene fragment, we analyzed and compared also genetic diversity among 190 bp of the hypervariable 327 bp gltA gene fragment extracted from our longer sequences and four reference sequences of Bartonella (Bartonella doshiae AF207827, B. grahamii Z70016, B. henselae L38987, and B. taylorii AF191502) from the GenBank database. The latter all contained the hypervariable 327 bp gltA gene fragment (La Scola et al. 2003, Gundi et al. 2009). Although the fragment that we compared was short (190 bp), sequence homology based on this region was only 90–95% (data not presented). Accordingly, these low similarity values for two genetic markers justify the description of two new species, supported also by phylogenetic analyses.
The similarity of our rpoB sequences with the human pathogenic B. elizabethae was only 88.30% in the case Candidatus Bartonella fadhilae n. sp. and 88.62% for Candidatus Bartonella sanaae n. sp. Moreover, analyses of our gltA sequences showed a similarity of 88.46% for Candidatus Bartonella fadhilae n. sp. with the pathogenic B. henselae and 88.80% similarity between Candidatus Bartonella sanaae n. sp. and B. henselae. Thus, any possible pathogenic consequences of infection with these new species cannot be currently assessed. Two pathogenic Bartonella spp. from humans in Africa are known to exist, B. henselae from Namibia (Noden et al. 2014) and B. quintana from Senegal (Diatta et al. 2014). Both these pathogenic Bartonella species have been reported also in South Africa with a high prevalence of up to 23% in people with HIV (Trataris-Rebisz 2012).
Our phylogenetic analyses of the rpoB sequences revealed four main genetic groups. In the first, the Candidatus Bartonella fadhilae n. sp. and Bartonella sanaae n. sp. sequences cluster with an unnamed Bartonella spp. from S. calurus from Egypt (Inoue et al. 2009), comprising a polyphyletic group/clade, supported by high bootstrap values, and a separate group/clade of other Bartonella sp. sequences from different regions of the world and derived from different hosts. This phylogenetic tree for rpoB sequences clearly highlights the difference between our sequences belonging to groups R1 and R2, and shows that these groups are separate from all other named Bartonella spp. With the exception of only one sequence of the pathogenic B. elizabethae, isolated from a human heart (Daly et al. 1993), all other Bartonella spp. sequences from this group/clade are derived from rodents.
Our phylogenetic analyses of gltA sequences also revealed four groups, the first comprising our Candidatus Bartonella fadhilae n. sp. and Candidatus Bartonella sanaae n. sp., with one sequence from D. dasyurus (sample H) from Egypt. This group clusters with B. florenciae from the greater white-toothed shrew C. russula from France and one unnamed Bartonella sp.; otherwise, this group contains different species of Bartonella from rodents from different regions of Europe.
The topology of the gltA tree differs from the rpoB tree primarily because of sample B, which belongs to group R2 of Candidatus Bartonella fadhilae n. sp. and is closely related to group G1 of Candidatus Bartonella sanaae n. sp. As reported previously by Paziewska et al. (2011), diversity in the gltA gene is highly associated with host specificity. Both samples in group G1 are from the same host species D. dasyurus and that is most likely the reason for sample B from D. dasyurus, identified as Candidatus Bartonella fadhilae n. sp, ending up so close to group G1 of Candidatus Bartonella sanaae n. sp. and why sample C from S. calurus identified as Candidatus Bartonella fadhilae n. sp. is separated from group G1 on this tree. This difference between samples B and C in the gltA gene sequence reflects also the difference that is apparent in the rpoB tree between the two variants of Candidatus Bartonella fadhilae n. sp.
An alternative interpretation of the differences between the two phylogenetic trees may be the possibility that horizontal gene transfer has occurred in the vector (flea), as reported in Paziewska et al. (2011). There is no recognized vector specificity for Bartonella spp. (Jusko 2015), and therefore, a horizontal gene transfer of the gltA gene is theoretically possible if a flea happens to be coinfected concurrently by two different species of Bartonella sp. from different hosts. As all the most similar gltA sequences (G1) originated from only one valley (W. Gebal), the possibility of a horizontal gene transfer of this marker cannot be excluded. Unfortunately, no other gltA sequences from the same or related host species are available from the GenBank database for comparison, in contrast to the rpoB gene, for which several sequences from S. calurus from Egypt have been deposited by Inoue et al. (2009) and are very similar to our sequences (although the exact region of origin in Egypt is not provided—the Bartonella strains in this study were isolated from rodents imported to Japan from Egypt, as pets). For these reasons, we consider the results of our phylogenetic analysis based on rpoB sequences to be more reliable, and based on this analysis, we have classified sample B from D. dasyurus as Candidatus Bartonella fadhilae n. sp.
Conclusions
Molecular and phylogenetic analyses of Bartonella samples from rodents from the Sinai Massif of Egypt have led to the description of two new species: Candidatus Bartonella fadhilae n. sp. and Candidatus Bartonella sanaae n. sp. The identification of their vectors, full microbiological assessment, and the medical significance of these species require further investigation.
Footnotes
Acknowledgments
Etymology: the Candidatus Bartonella fadhilae n. sp. is named after the engineer Fadhil Naqi (the father of the first author of this study, M.A.), who was a fatal victim of terrorism in Iraq in 2005. The Candidatus Bartonella sanaae n. sp. is named after the dentist Sanaa Shukur (the mother of the first author of this study, M.A.), who took care of the family following loss their father. This study was funded by the National Science Center (NCN), Poland, grant OPUS 2011/03/B/NZ6/02090 (2012–2015) (to A.B.) and by the Ministry of Science and Higher Education through the Faculty of Biology, University of Warsaw intramural grant DSM number 140000/501/86-110101 (2014/2015) (to M.A.).
Authors' Contributions
A.B. designed the study and supervised laboratory and field analyses, M.A., R.W.F., and L.D. performed molecular and phylogenetic studies, M.B. participated in biological characterization of novel Bartonella spp., F.G. and S.Z. organized and supervised field work in Sinai, E.J.M., J.B.B., E.M.M., and J.M.B. carried out the laboratory and field studies and contributed to the drafting of the manuscript. All authors read and approved the final version of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.