
Abstract
Here we present a real-time one-step reverse transcriptase–polymerase chain reaction SYBR Green assay derived from the method reported by van Elden and collaborators (2001) that ensures the rapid, sensitive, and cost-effective detection of both seasonal influenza A virus and emerging (H1N1) swine-origin influenza A virus (S-OIV). In addition to this screening test, which successfully detected both seasonal influenza A virus and S-OIV in human clinical samples, we showed that the probe initially designed by van Elden and collaborators could detect seasonal influenza A virus, but not S-OIV; a new probe was designed and tested that specifically detects S-OIV, but not seasonal influenza A. Both probe-based assays were validated by testing human clinical samples and specifically detected either seasonal influenza virus or S-OIV. Finally, in silico analysis of databases predicted that minor modifications of the van Elden primers would facilitate the use of this assay for the broad spectrum detection of all currently characterized variants of influenza A virus, including avian strains.
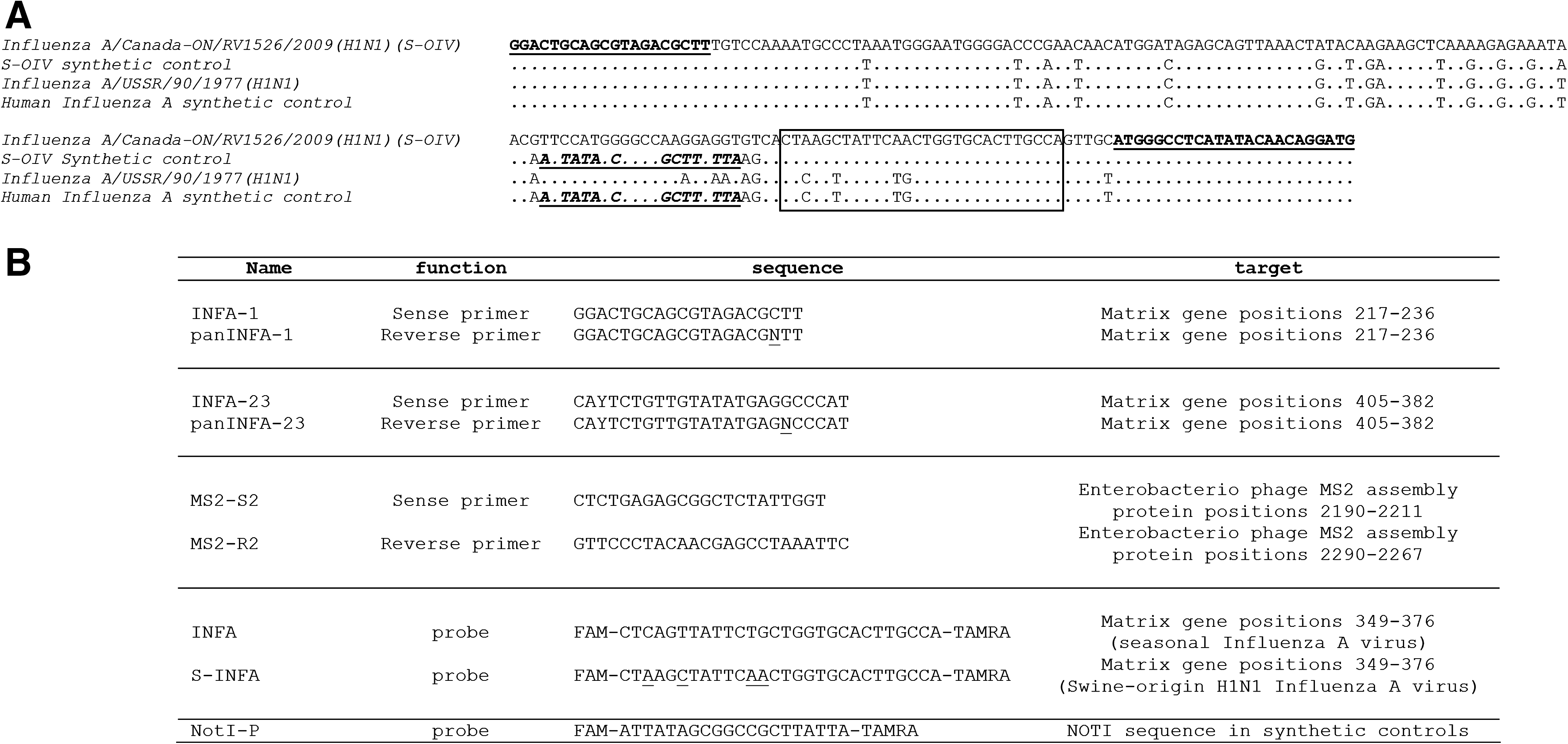
Since 2002, we have been using a real-time PCR assay designed by van Elden and collaborators (2001) for the routine molecular detection of influenza A virus. In this test, a 189 nt fragment within the Matrix gene of the virus is amplified using the INFA-1 and INFA-23 primers and hybridized with the INFA-specific probe (see Fig. 1) using a two-step RT-PCR protocol (i.e., a reverse transcription reaction using random hexaprimers, followed by a real-time PCR reaction). This assay has proved to be robust and able to detect human influenza virus subtypes H1N1 and H3N2 in clinical samples. Moreover, it can be combined with the simultaneous detection of influenza B virus in a multiplex assay (van Elden et al. 2001).
(
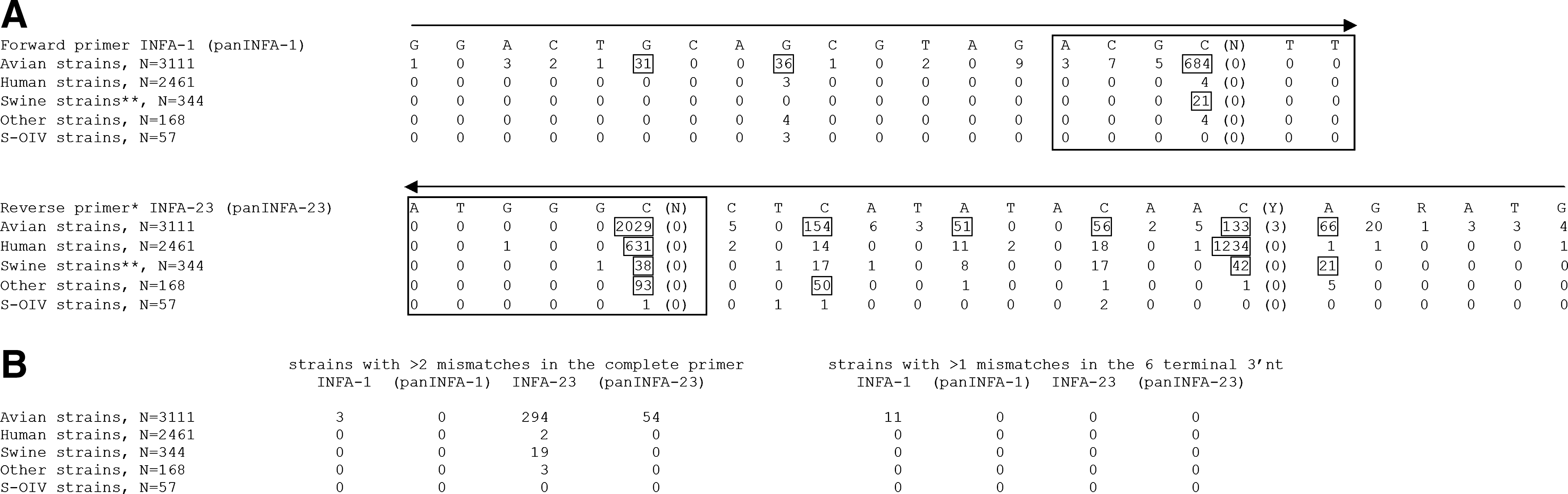
The recent spread of the H1N1 swine-origin influenza A virus (S-OIV) led us to reconsider this assay. The Matrix gene of S-OIV is of porcine origin, and genetic analysis of sequences available in GenBank revealed an ∼13% nt divergence between the region targeted by the assay in the new porcine H1N1 virus and in previously circulating H1N1 or H3N2 human strains. In particular, the region chosen for probe hybridization includes four nt mismatches (see Fig. 1), suggesting that the probe initially designed by van Elden and collaborators would not efficiently detect the new porcine virus. In contrast, the van Elden primers matched the corresponding sequence of the new virus (Figs. 1 and 2).
(
Accordingly, the primers from the van Elden assay were used to elaborate a simple SYBR Green one-step real-time RT-PCR assay as described hereafter. Extraction: 200 μL of naso-pharyngeal sample was spiked with 5 μL of MS2 phage suspension and used for nucleic acid extraction (elution volume: 90μL) using the EZ1 Biorobot and the virus mini kit (both from Qiagen, Courtaboeuf, France). Amplification: one-step RT-PCR gene amplification was performed in a 25 μL final volume, using 5 μL of RNA extract, 1.4 μL of each 10 μM primer solution, and the one-step QuantiTect SYBR Green RT-PCR Kit master mix with Uracil-N-Glycosylase (UNG) (Qiagen).
Two distinct reactions were conducted using the Mx3005P® QPCR System thermocycler (Stratagene-Agilent, Massy, France) and the following thermal profile. Segment 1: 50°C, 2 min; segment 2: 95°C, 15 min; segment 3: 94°C, 15 s; 60°C, 30 s; and 72°C, 30 s (45 × ), followed by the analysis of a thermal dissociation curve. One reaction included the INFA-1/INFA-23 primer set for the detection of the influenza A virus; the other included the MS2-S2/-R2 primer set for detection of MS2 phage detection (see Fig. 1B for a list of primers and probes used in the current study). The latter reaction was used as an internal control for monitoring the extraction and RT-PCR steps, and in particular the detection of amplification inhibitors in the biological material tested (see the dissociation curve in Fig. 3, with a Tm at 82°C).
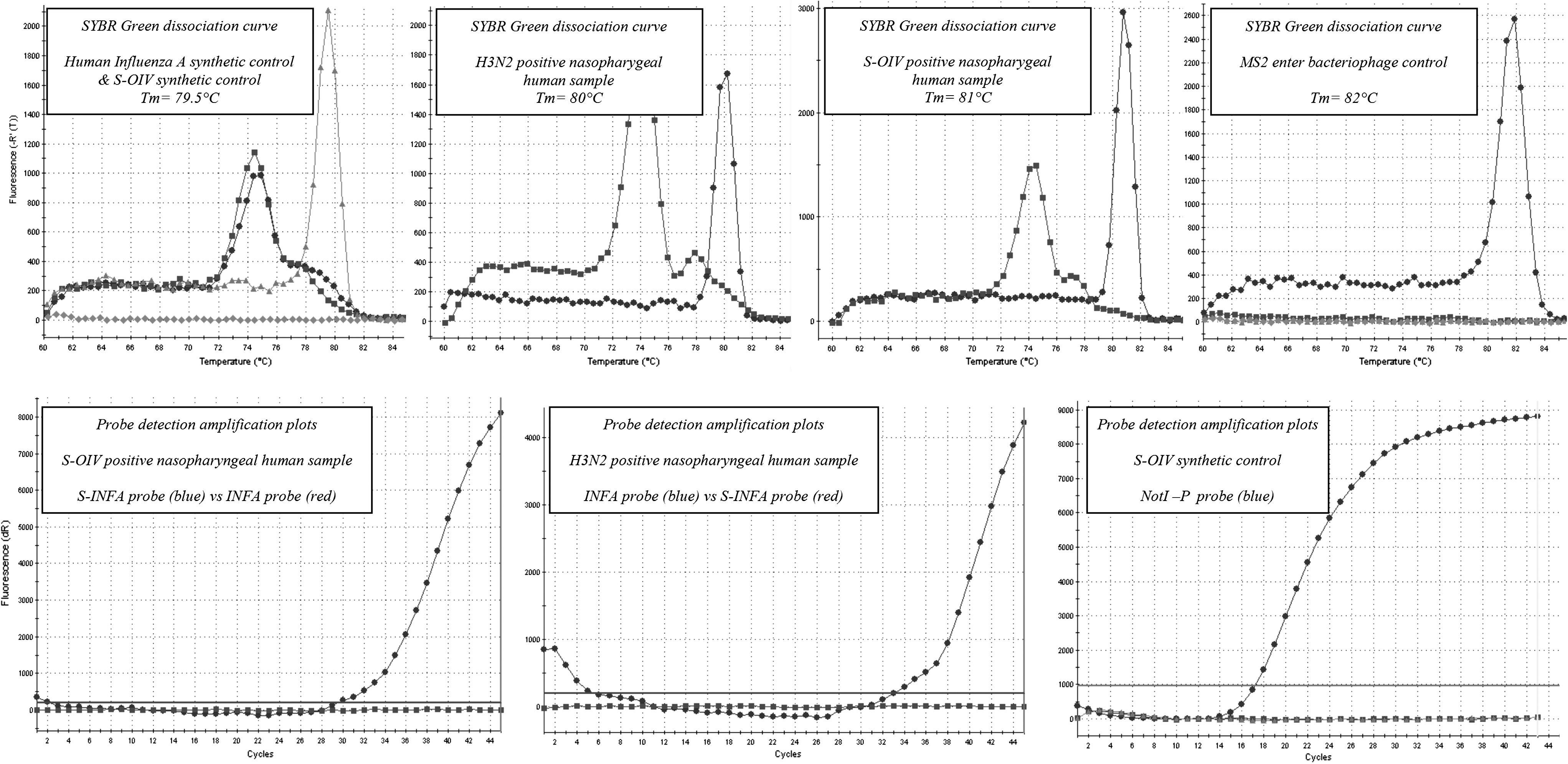
Real-time reverse transcriptase–polymerase chain reaction amplification of synthetic controls and clinical samples. First line shows SYBR dissociation curves for influenza virus synthetic controls, H3N2-positive and S-OIV–positive clinical samples, and MS2 control. Second line shows specific probe detection of H3N2 and S-OIV in clinical samples using INFA and S-INFA probes that of S-OIV synthetic control using NotI-P probe (system designed for the detection of incidental contamination by synthetic controls).
In addition, synthetic RNA transcripts (including a specific extrinsic specific sequence, that is, a NotI restriction site; see Fig. 1A) were produced by amplification with a primer containing the T7 promoter sequence and by transcription with the T7-MEGA shortscript (Ambion, Applied Biosystems, Courtaboef, France) according to the manufacturer's instructions. They were quantified by spectrophotometry and used as positive controls for RT-PCR amplification.
Synthetic RNA controls were detected with a Tm of the amplicons at 79.5°C (see Fig. 3). When serial dilutions were tested, 50 RNA copies per reaction could be reproducibly and unambiguously detected (equivalent to ∼4.5 viral RNA copies per μL of clinical sample tested; data not shown). With regard to these performances, the assay was subsequently used to detect genomic viral RNA in clinical samples. It was able to detect either classicalinfluenza A virus (see Fig. 3, detection of H3N2 virus in a human naso-pharyngeal sample, with a Tm at 80°C) or the newly emerging H1N1 porcine virus (detection of S-OIV in a human naso-pharyngeal sample, with a Tm at 81°C), justifying its use for the immediate detection of the influenza A virus in clinical samples.
In addition to this test, one-step real-time RT-PCR assays were performed using either the probe designed by van Elden and collaborators, or the equivalent probe (S-INFA) designed from the sequences available for S-OIV (see Fig. 1) and the following protocol. Amplification: Genetic amplification was performed in a one-step RT-PCR format and a 25 μL final volume, using 5 μL of RNA extract, 1 μL of each 10 μM primer solution, 0.4 μL of 10 μM probe solution, and the Super Script III Platinum One step qRT PCR System (Invitrogen, Cergy Pontoise, France). Two reactions were conducted using the Mx3005P QPCR System thermocycler and the following thermal profile. Segment 1: 48°C, 30 min; segment 2: 95°C, 2 min; segment 3: 95°C, 15 s; 60°C, 1 min (45 ×). One reaction included the INFA-1/INFA-23 primer set and the INFA probe for the detection of the seasonal influenza A virus; the other included the same primers and the S-INFA probe for the detection of S-OIV. Synthetic RNA transcripts specific for human seasonal influenza A virus and S-OIV were used as positive controls for RT-PCR amplification.
Using both synthetic RNA transcripts and human samples positive for either H3N2 or S-OIV, it could be shown (see Fig. 3) (i) that the van Elden and S-OIV probes could efficiently detect the H3N2 and S-OIV, respectively, but (ii) that the van Elden probe could not detect S-OIV, while the S-OIV probe could not detect seasonal influenza virus. A probe designed to detect specifically the synthetic positive control (NotI-P) was used to rule out the possibility of accidental amplification of the control.
Altogether, these results suggest that this SYBR Green one-step real-time RT-PCR assay is a simple, inexpensive, but very effective tool for screening clinical samples for the presence of influenza A virus. Most importantly, positive samples may be secondarily tested using the same primers and the van Elden (INFA) and S-OIV (S-INFA) probes to discriminate between seasonal and S-OIV. It should also be noted that the direct sequencing of the products resulting from the SYBR Green assay produced the definitive differentiation of seasonal and S-OIV strains.
In addition to this initial study, which provides a rapid and pragmatic solution to the detection of the newly emerging S-OIV, we analyzed in depth: (i) 6084 influenza A Matrix sequences available in the Influenza Virus Resource database ( (ii) 57 S-OIV influenza A Matrix sequences available in GenBank.
The performance of the van Elden primers was evaluated in silico for each group of influenza A viruses (see Fig. 2). This analysis revealed that these primers are indeed highly appropriate for the detection of human seasonal influenza A virus and for most porcine strains, including the newly emerging S-OIV. However, minor but important modifications among the six 3′ terminal nucleotides of each primer would ensure the detection of viruses from other groups, in particular avian viruses (see Fig. 2B). Modified primers (panINFA-1 and panINFA-23) were predicted to be capable of detecting all Matrix sequences available to date, including strains of avian (including H5N1 viruses) and porcine (including H1N1 viruses) origin, irrespective of hemagglutinin and neuraminidase subtypes.
In conclusion, we propose a simple and cost-effective amplification protocol for the detection of both seasonal influenza virus and newly emerging S-OIV strains. Its effectiveness relies on tried and tested amplification primers that were shown to be extremely reliable for the screening of clinical samples and on actual detection of both seasonal and H1N1 S-OIV in human clinical samples. In silico analysis predicts that minor modifications of these primers would allow the use of this assay for the broad spectrum detection of all currently known variants of influenza A virus, including avian strains.
Disclosure Statement
No competing financial interests exist.