
Abstract
Background:
The liver and thyroid have complicated effects on each other’s functions and metabolic homeostasis in the body. Both hypo- and hyperthyroidism influence hepatic carbohydrate and fat metabolism to regulate circulating glucose, cholesterol, and triglyceride levels. Hypothyroidism and “intrahepatic” hypothyroidism also contribute to the development of hypercholesterolemia and metabolic dysfunction-associated steatotic liver disease (MASLD). Likewise, hepatic dysfunction can modulate thyroid function by reducing thyroid hormone (TH) concentrations and their effects on peripheral tissues.
Summary:
In this review, we examine the impact of thyroid disorders and their treatment on hepatic physiology, metabolism, and pathology, as well as the influence of liver disease on thyroid function. We also describe the clinical and experimental evidence for THs playing significant roles in metabolic conditions such as metabolic syndrome, hyperlipidemia, and MASLD. Additionally, we summarize the current literature on the use of thyromimetics for the treatment of metabolic diseases.
Conclusions:
Recognizing the effects of the thyroid and THs on hepatic metabolism and fuel utilization, and the liver’s role in modulating systemic TH action, is important for optimal clinical management of patients with thyroid and/or liver diseases. New emerging concepts on TH actions in the liver and the efficacy of thyromimetics for the treatment of MASLD have reshaped our understanding of the thyroid–liver relationship and the roles of THs in the pathogenesis and treatment of metabolic diseases.
Introduction
The relationship between the liver and the thyroid gland is dynamic and complicated. Although much is known about the effects of thyroid hormones (THs) on the metabolism of other tissues and the critical roles of the liver in nutrient metabolism, the direct and indirect interactions between them have often been overlooked despite their important roles in normal physiology and diseases that affect both tissues. Hypo- and hyperthyroidism have profound effects on carbohydrate and fat metabolism in the liver and affect cholesterol and triglyceride (TG) homeostasis in the circulation and in the liver. Hepatic dysfunction can also affect circulating free TH concentrations and their effects on peripheral tissues. Additionally, autoimmune hepatic and thyroid disease can occur concurrently. Given the cross talk between these two important organs, regular monitoring of both thyroid and liver function is important when managing patients with conditions that affect one or both organs.
In this review, we will cover the effects of thyroid disease on the liver, and vice versa. We will also examine the role of THs on metabolic conditions such as hyperlipidemia and metabolic dysfunction-associated steatotic liver disease (MASLD). Better understanding of the interactions between the thyroid and liver will lead to improved management of patients with conditions involving either or both of these tissues.
Mechanisms of TH Action
Hypothalamus, pituitary, and thyroid axis regulation of serum TH concentration
Serum concentrations of THs (T3, triiodothyronine; T4, thyroxine) are regulated by a negative feedback system involving the hypothalamus, pituitary, and thyroid (HPT) axis. The hypothalamus produces thyrotropin-releasing hormone (TRH), a tripeptide that prompts the pituitary to secrete thyrotropin (TSH). In turn, TSH stimulates the thyroid gland to produce and release THs into the bloodstream. Importantly, elevated levels of THs inhibit both TRH and TSH secretion, to ensure coordinated negative regulation of the HPT axis.
The thyroid gland actively takes up iodide from the bloodstream via the sodium/iodide symporter located on the basolateral surface of thyrocytes. These cells are arranged as follicles with a central colloid zone. Once inside the cell, iodide is transported into the colloid zone by the pendrin protein (encoded by the SLC26A4 gene), located on the apical membrane. Here, iodide is linked to tyrosine residues on thyroglobulin by dual oxidase and thyroid peroxidase (TPO) enzymes, to form mono-iodotyrosine (MIT) and di-iodotyrosine (DIT). These MIT and DIT molecules are then coupled by TPO to generate the T3 and T4, which are stored within the colloid. 1,2
After TSH signaling, thyroglobulin is internalized at the apical surface of the thyrocytes and undergoes degradation to release both T3 and T4 into the circulation via the monocarboxylate transporter (MCT8). Although most of the secreted TH is in the form of T4, T3 is the more active form, with ∼10 times higher potency. Most circulating T3 and T4 are bound to carrier proteins secreted by the liver, such as thyroxine-binding globulin (TBG), transthyretin (TTR), and albumin (HSA), acting as hormone reservoirs. 3 These protein-bound hormones are inactive so there is a dynamic equilibrium between the bound and free (active) forms of T4 and T3 (fT4 and fT3) in the bloodstream. Circulating fT3 and fT4 then enter target tissues via plasma membrane transporters with MCT8 and the organic anion transporting peptide, OATP1C1, as the main hepatic TH transporters.
Regulation of intracellular and extracellular TH concentrations by deiodinases
Deiodinases (DIOs) comprise a family of selenoproteins that play a crucial role in maintaining both circulating and intracellular concentrations of T3, T4, and their metabolites. 4 These enzymes fine-tune the biological activity of THs by modulating the conversion and inactivation of THs. DIO1 and DIO2 convert T4 to the more active T3, while DIO3 inactivates T3 by converting it to 3,3′-diiodo-l-thyronine (T2) and forms reverse T3 (rT3) from T4. Both DIO1 and DIO2 also mediate the conversion of rT3 to T2. The expression of these DIOs varies by tissue, with DIO1 being abundant in the liver and DIO2 in brown adipose tissue, skeletal muscle, and the brain. Interestingly, the activity of DIO1 can be inhibited selectively by β-blockers, propylthiouracil (PTU), and iodine, whereas DIO2 is unaffected.
The liver regulates the conversion of T4 to T3 to control the amount of active TH in both the circulation and the liver. This conversion is mediated by DIO1 and DIO3, which convert T3 to inactive T2. Interestingly, DIO1 expression and activity are also induced by T3 to create a positive feedback mechanism. The liver also modifies T2 into metabolites that are released into the gut and cleared from the body. Finally, the liver regulates the amount of free (unbound) circulating THs that are available to the liver and other tissues by virtue of its synthesis of serum binding proteins (see the section “Hypothalamus, Pituitary, and Thyroid Axis Regulation of Serum TH Concentration”).
Gene transcription by TH
There are two major TH receptors, THRα and THRβ (also called THRα1 and THRβ1, respectively, in humans) that are encoded on two separate genes: THRA and THRB. 2,5 THRs are ubiquitously expressed, but their relative distribution varies among different tissues. THRα is predominantly expressed in the heart, bone, skeletal muscle, gastrointestinal tract, and brain, whereas THRβ1 is widely distributed but is the major THR isoform in the liver and kidney. It is estimated that THRβ isoform comprises 80% of the THRs in the liver. 6
THRs belong to a large family of nuclear receptors that regulate gene transcription in a hormone-dependent manner and thus can be categorized as “ligand-regulatable” transcription factors. 7 Both THRα and THRβ have high protein sequence homology and appear to regulate the same target genes. However, it remains possible that there may be THR isoform-specific transcriptional regulation of a limited number of genes, particularly during development when THRα is expressed earlier than THRβ. THRs regulate target gene expression both positively and negatively. 8,9 Details on the mechanism of THR complexes regulating the gene transcription of target genes via interactions with coactivators and corepressors, as well as nongenomic actions of THRs, are described elsewhere. 8,10
Hypothyroidism Effects on Liver Function
Causes of hypothyroidism
Most cases of hypothyroidism are classified as primary hypothyroidism, arising from the failure of the thyroid gland to produce sufficient THs. The most common cause of hypothyroidism is the autoimmune disease, Hashimoto’s thyroiditis, which has a high overall prevalence in the adult population of 7.5%; however, there is wide variation depending upon geographical location and socioeconomic status of patients. 11 The course of disease is slow and insidious, leading to overt hypothyroidism if undiagnosed and untreated. Other major causes of hypothyroidism include iatrogenic due to overtreatment of hyperthyroidism, goitre, or thyroid cancer by drugs, radioiodine (RAI), or surgery; iodine deficiency; and thyroiditis from drugs, postpartum, or viral illnesses such as COVID-19. Rarely, extensive pituitary disease can lead to decreased TSH secretion and secondary hypothyroidism. These conditions are described in detail elsewhere and are not the subject of this review. 12,13 However, it should be emphasized that the effects of hypothyroidism on the liver are similar regardless of the cause of the thyroid disease.
Overlapping clinical features of hypothyroidism and liver disease
Clinical features of subclinical hypothyroidism and early hypothyroidism may overlap with those manifested by mild liver disease as both exhibit relatively nonspecific symptoms such as fatigue, weakness, myalgia, mental status change, and muscle cramping. As both conditions progress, dyspnea on exertion, altered mental status, peripheral edema, and pericardial effusion can occur in both severe hypothyroidism and liver failure. Severe hypothyroidism has been associated with hyperammonemia, particularly in patients with underlying liver dysfunction. 14,15 Indeed, hypothyroidism as a cause of hepatic encephalopathy has been reported in patients with hepatitis C virus–related cirrhosis. 14,16 Several cases of myxedema coma and hyperammonemia also have been reported. 14,16 –18 In these cases, patients typically presented with hyperammonemic coma that did not respond to conventional treatment for hepatic encephalopathy, and improved only after TH replacement. 18
The precise mechanism(s) for how severe hypothyroidism causes hyperammonemia or potentiates the hyperammonemia caused by preexisting liver disease remains unclear. In a rodent model, hypothyroidism is associated with decreased glutamine synthetase expression in the liver. 19 Additionally, decreased urea synthesis rate is found in women with hypothyroidism, 20 suggesting there is a decrease in ammonia clearance during hypothyroidism. Decreased gut motility favoring bacterial production and absorption of ammonia, particularly when combined with reduced hepatic glutamine synthetase activity, also may contribute to hyperammonemia. 19 Additionally, the proteolysis and release of increased amounts of amino acids from skeletal muscle can further add to hepatic ammonia overproduction. Finally, secondary causes such as heart or renal failure may indirectly affect serum ammonia levels by decreasing metabolism and clearance. Myxedema ascites is an uncommon complication of severe hypothyroidism and is thought to be due to decreased lymphatic drainage 21 and altered capillary permeability. 22 The ascites usually resolves after TH replacement.
Elevated serum levels of hepatic enzymes such as alanine aminotransferase (ALT) and gamma glutamyl transferase (GGT) concentrations are observed in patients with either hypothyroidism or hepatitis due to primary liver disease. Increases in serum aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) levels also occur in hypothyroidism and may be due to myopathy induced by low circulating TH levels. 23,24
Clinical association between hypothyroidism and MASLD
Initial meta-analyses examining the association of hypothyroidism and subclinical hypothyroidism with MASLD revealed conflicting results; however, subsequent studies strongly suggest that both hypothyroidism and subclinical hypothyroidism are associated with MASLD. 25 –28 A genome-wide association study shows high correlation between hypothyroidism and MASLD, with odds ratio (OR) of 1.7578 (confidence interval [CI 1.1897–2.5970], p = 0.0046). 29 A meta-analysis also demonstrates serum TH and TSH levels have negative and positive correlations with the risk for MASLD, respectively. 30 In particular, the incidence of liver fibrosis is significantly higher in individuals with high TSH levels. The findings of five major meta-analyses of this relationship between hypothyroidism and MASLD are summarized in Table 1. Although it appears that the degree of hypothyroidism correlates with the severity of MASLD, it is possible that TSH itself increases the risk for MASLD independent from decreased serum TH levels (see the section “TSH Regulation of Liver Function”). Analyses of individual data from 14,797 adult participants aged 20–74 in the National Health and Nutrition Examination Survey (NHANES) III study show there is a “dose–response” relationship between TSH and MASLD. 31 TSH levels >2.5 IU/mL are associated with a 1.5-fold increase in the risk of MASLD and a 1.8- to 2.5-fold increased risk of liver fibrosis, based upon adjustments for confounding variables. 32 This observation is also consistent with previous studies showing that the severity of hepatic steatosis, metabolic dysfunction-associated steatohepatitis (MASH) activity, and fibrosis inversely correlates with thyroid function. 33,34
Meta-Analyses Evaluating the Link Between Metabolism-Associated Steatotic Liver Disease and Hypothyroidism
aOR, adjusted odds ratio; CI, confidence interval; HR, hazard ratio; I 2, heterogeneity statistic (% of variation due to heterogeneity); MASLD, metabolic dysfunction-associated steatotic liver disease; OR, odds ratio; T3, triiodothyronine; T4, thyroxine; TSH, thyrotropin.
Patients with overt hypothyroidism typically exhibit high total cholesterol (TC), high low-density lipoprotein cholesterol (LDL-C), normal high-density lipoprotein cholesterol (HDL-C), normal-to-borderline high TGs, normal apolipoprotein A1 (apoA1), high apolipoprotein B (apoB), and normal-to-high lipoprotein(a) [Lp(a)] levels, whereas patients with subclinical hypothyroidism display borderline high TC, LDL-C, and apoB but desirable HDL-C, TG, apoA, and Lp(a) levels. 35 Treatment of hypothyroid and subclinical hypothyroid patients with levothyroxine (LT4) leads to improvements in their lipid profiles. 35 –37 These clinical features resemble those found in patients with MASH since the latter typically have high serum TC, LDL-C, high apoB, and elevated TG levels due to metabolic defects in lipid metabolism. Hypothyroidism, in combination with insulin resistance from obesity and diabetes, frequently occurs in patients with MASLD and exacerbates hepatic TG accumulation and hepatic insulin resistance. 38
Effects of hypothyroidism on hepatic metabolism and development of MASLD
Hypothyroidism causes profound changes in hepatic lipid, glucose, and protein metabolism. 39 Hypothyroidism leads to elevated serum cholesterol by several major mechanisms including increased cholesterol absorption in the small intestines and liver mediated by the Niemann–Pick C1-like 1 protein; decreased expression of cell surface LDL-C receptors leading to reduced plasma LDL-C clearance and increased circulating apoB lipoproteins; reduced plasma cholesteryl ester transfer proteins (CETPs), which shift serum cholesterol-carrying species from HDL-C to LDL-C and very low-density lipoprotein (VLDL); decreased reverse cholesterol transport and bile acid synthesis to reduce hepatic cholesterol clearance; and decreased expression of microRNA 181 (miR181) causing increased expression of sterol o-acyltransferase 2 (SOAT2), an enzyme that generates cholesterol esters that are packaged in lipoproteins. 40 –42 Hypertriglyceridemia is caused by decreased plasma TG clearance due to reduction in lipoprotein lipase levels and increased production of VLDL to mitigate hepatic lipotoxicity due to downregulated autophagy of fat droplets and reduced expression of carnitine palmitoyltransferase-1a (CPT1a), the rate-limiting enzyme in fatty acid oxidation during hypothyroidism.
Hypothyroidism also likely contributes to the development of MASLD through its effects on lipid metabolism, insulin resistance, inflammation, and oxidative stress. In particular, patients with hypothyroidism have impaired cholesterol metabolism and clearance, decreased β-oxidation of fatty acids, and aberrant fatty acid synthesis in the liver. TH also plays a critical role in the autophagy of lipids from fat droplets (lipophagy) and β-oxidation of fatty acids by inducing the gene expression of autophagy proteins and CPT1a and acyl-CoA dehydrogenases. Moreover, TH increases mitochondrial biogenesis and mitophagy to maintain mitochondrial quality control and reduce the production of reactive oxygen species (ROS). 2,43 Inhibition of these processes during hypothyroidism initially causes TG and cholesterol accumulation in the liver. 43 However, as disease progresses, the accumulation of saturated long-chain fatty acids within the liver eventually overwhelms its capacity to metabolize and clear them. These excess saturated long-chain fatty acids can generate toxic metabolites such as ceramides and dihydroxy glycerols that cause lipotoxicity, inflammation, and cell death. 43 These inflammatory changes induce activation and transformation of hepatic stellate cells into fibroblasts to cause fibrosis in MASH 43,44 Moreover, insufficient metabolism of FFAs leads to hepatosteatosis and lipotoxicity that gives rise to inflammation and fibrosis in the liver 41,45 (Fig. 1). The latter mechanisms are thought to play key roles in the development of MASLD during hypothyroidism and in the pathogenesis of de novo MASLD and are the basis for the beneficial effects of TH and thyromimetic drugs as therapies for MASH. Importantly, T4, T3, and its natural metabolite, T2, can all increase hepatic lipophagy and lipid catabolism to reverse the hepatic and circulating lipid abnormalities. 39,46
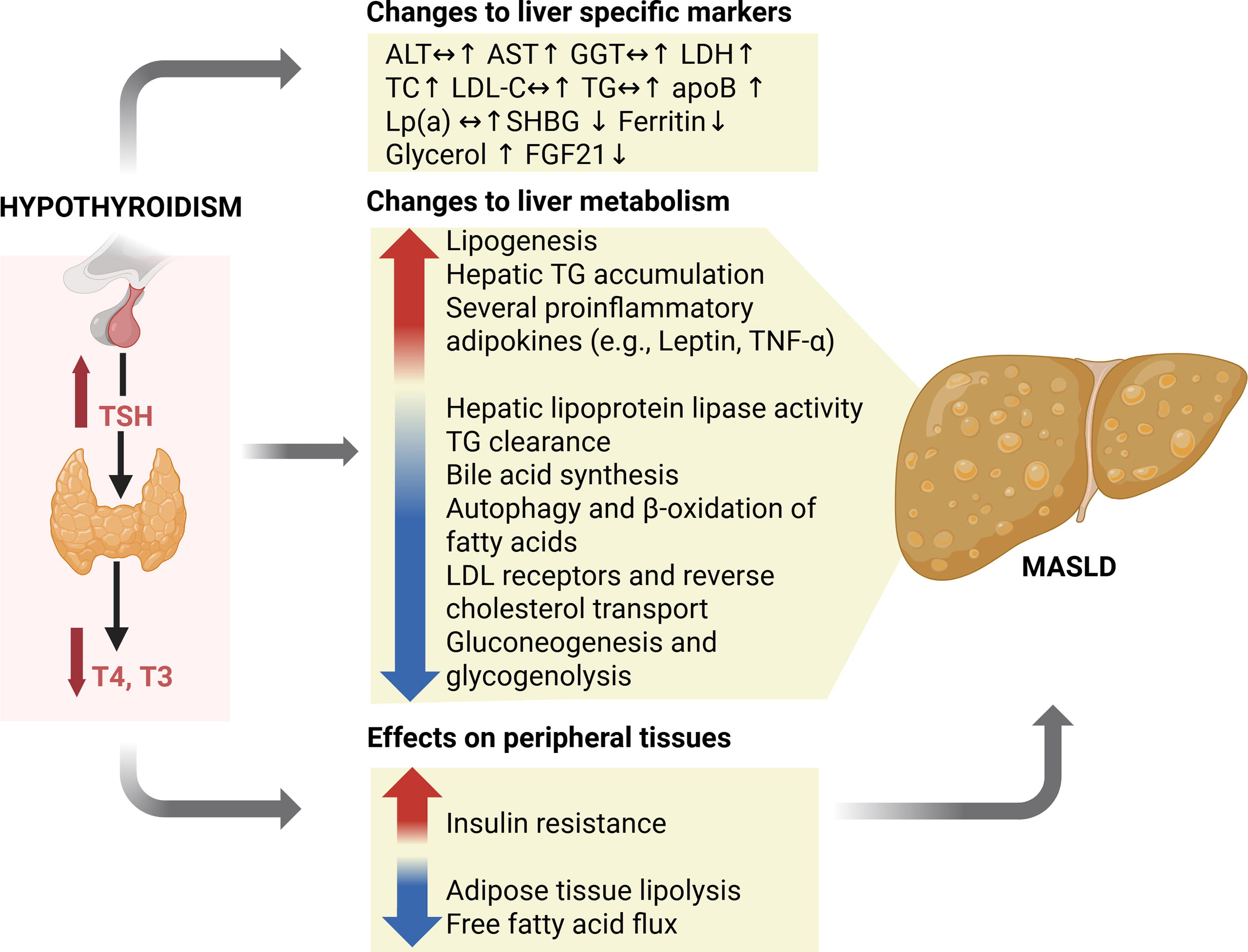
Effects of hypothyroidism on liver metabolism contributing to the development of MASLD and changes in liver-specific markers (Image created using https://BioRender.com). ALT, alanine aminotransferase; apoB, apolipoprotein B; AST, aspartate aminotransferase; GGT, gamma glutamyl transferase; FGF21, fibroblast growth factor 21; LDH, lactate dehydrogenase; LDL-C, low-density lipoprotein cholesterol; Lp(a), lipoprotein(a); MASLD, metabolic dysfunction-associated steatotic liver disease; SHBG, sex hormone-binding globulin; TC, total cholesterol; TG, triglycerides; TNF-α, tumor necrosis factor alpha. ↔ No change ↑ Increased ↓ Decreased.
Hypothyroidism also increases the expression of lipogenic enzymes. 47 LT4 replacement therapy reverses these effects and increases insulin sensitivity as reflected by decreased glucose-stimulated insulin secretion. 48 Furthermore, a recent study also suggests that TH decreases hepatic CYP8B1 expression to increase bile acid secretion. 49 This, in turn, represses Farnesoid X receptor (FXR) signaling in the gut and potentiates glucagon-like peptide-1 (GLP-1) secretion, resulting in enhanced insulin secretion. 49 The opposite effects are expected to occur in hypothyroidism. Therefore, these exciting data suggest that the gut in tandem with the liver may be a major site responsible for the glucose- and lipid-lowering actions of TH. 49
Hypothyroidism causes downregulation of gluconeogenic enzymes, glucose-6-phosphatase and phosphoenolpyruvate carboxykinase, and glycogenolytic enzymes, phosphorylase kinase and glycogen phosphorylase, to reduce liver glucose output that can lead to hypoglycemia. 50,51 Hypothyroidism also reduces the gene expression of β2-adrenergic receptor and increases the expression of inhibitory G protein of the adenylate cyclase cascade to blunt the effects of epinephrine and glucagon. 8 Reduced central effects of TH on the sympathetic pathway linking the paraventricular hypothalamus to the liver can also reduce hepatic glucose production. 52 Additionally, the downregulation of the gluconeogenic enzymes leads to higher de novo lipogenesis. 43 Notably, hypothyroidism leads to insulin resistance by reducing the sensitivity of skeletal muscle and adipose tissue to insulin, thereby impairing glucose disposal. 53 This effect partially offsets the reduction in serum glucose levels that would otherwise result from decreased hepatic gluconeogenesis and glycogenolysis.
Hypothyroidism is associated with increased circulating levels of proinflammatory adipokines, which may contribute to hepatic inflammation and fibrosis. Furthermore, oxidative stress during hypothyroidism can worsen cellular injury and insulin resistance due to diminished fatty acid β-oxidation and increased lipid peroxidation. 54 Of note, these metabolic effects of hypothyroidism mirror those occurring in MASLD. Figure 1 summarizes the effects of hypothyroidism on liver metabolism contributing to the development of MASLD and changes in liver-specific markers.
Apart from its complex effects on lipid and glucose metabolism, THs exert other metabolic actions on the liver. Several liver tissue-specific markers correlating with human thyroid status have been proposed. 55 Sex hormone-binding globulin (SHBG) and ferritin have been extensively studied as potential liver-specific tissue biomarkers for TH status and can be measured by routine biochemical assays available in most clinical laboratories. TH increases hepatic gene expression of SHBG indirectly by inducing the expression of hepatocyte nuclear factor 4 alpha (HNF-4α), a nuclear transcription factor to increase synthesis and secretion of SHBG into the circulation. 56 Accordingly, SHBG levels are either reduced or normal in hypothyroidism. 57 Recently, it was reported that serum SHBG concentrations in patients with MASH predicts the efficacy of a liver- and THRβ-selective agonist, Resmetirom, to reduce hepatic steatosis. 58 These findings suggest that measurement of serum SHBG concentrations has potential clinical utility as a noninvasive marker of TH action before and during the treatment of MASH. 58
T3 directly induces hepatic ferritin gene expression and increases iron regulatory protein binding to the iron-responsive element-binding activity of ferritin messenger RNA (mRNA) to increase its translation. 59 Therefore, ferritin levels decrease in hypothyroidism and increase after TH treatment. However, factors such as body mass index, insulin resistance, age, use of oral contraceptives, and pregnancy can influence SHBG levels, whereas inflammation and iron depletion affect the measurement of ferritin levels, so these factors need to be considered when using ferritin as a marker of TH action in the liver. 55 Other potential hepatic biomarkers for hypothyroidism include glycerol and fibroblast growth factor 21, but their clinical utility has not been established yet. Elevated serum LDL-C, high TGs, and TC levels are frequently found in hypothyroidism, but their measurements lack specificity due to contributions from many other tissues besides the liver.
Recently, metabolomic profiling has been used to identify specific serum metabolic biomarkers and pathways that distinguish patients with newly diagnosed subclinical hypothyroidism and overt hypothyroidism from healthy controls. 60 In particular, metabolite levels from cholesterol metabolism, primary bile acid biosynthesis, bile secretion, and steroid hormone biosynthesis pathways are perturbed in patients with hypothyroidism and subclinical hypothyroidism compared with normal controls. This study raises the exciting possibility that detection of abnormal serum levels of metabolites may someday be useful for diagnosing subclinical and overt hypothyroidism 60 ; however, further studies are needed to determine its utility.
Hypothyroidism and metabolic syndrome
Metabolic syndrome (MetS) is composed of a group of interconnected metabolic abnormalities including central obesity, elevated serum TG and LDL levels, decreased serum HDL-C level, hypertension, and hyperglycemia. MASLD is frequently considered a hepatic manifestation of MetS and linked with other metabolic risk factors such as type 2 diabetes mellitus (T2DM), hyperlipidemia, coronary artery disease, and chronic kidney disease, which contribute to increased risk for all-cause mortality. 61,62 Accordingly, these other complications, which are hallmarks of MetS, can also be considered to be extrahepatic manifestations of MASLD. Besides the high co-occurrence of MetS in hypothyroidism, it is noteworthy that hypothyroidism and subclinical hypothyroidism also frequently occur concomitantly with MetS. Notably, one report showed that thyroid dysfunction occurred in 32% of patients with MetS. 63
Apart from its relationship to MASLD, TH also contributes to the other aspects of MetS by virtue of its diverse physiological and metabolic actions on glucose and lipid homeostasis, appetite control, adiposity, weight, and blood pressure. 64 These systemic effects, particularly on glucose and lipid regulation, are highly regulated by the liver. In addition to changes in serum lipid profiles found in patients with hypothyroidism, there is a higher prevalence of T2DM in patients with hypothyroidism compared with the general population, with factors such as age, gender, obesity, presence of TPO antibody, and prior hospitalization, also contributing to the increased prevalence. 65 –67 Additionally, a progressive decline in thyroid function, even within the euthyroid range, is associated with an increased risk of developing T2DM, and this relationship is observed regardless of gender or thyroid autoimmunity. 68 In a population-based study using NHANES III data, there is a strong correlation between MASLD progression with increasing plasma TSH levels that is also associated with an increase in all-cause mortality, primarily due to cardiovascular complications. 34 This study showed that serum TSH level is independently associated with MASLD progression at different levels ranging from hypothyroidism to euthyroidism.
“Intrahepatic hypothyroidism” in MASH
DIOs regulate the intrahepatic TH concentrations. High levels of DIO1 and low levels of DIO3 are found in normal adult hepatocytes. In mouse models of MASLD, DIO1 expression and activity increase as a compensatory mechanism during hepatosteatosis to maintain normal intracellular T3 levels. 69 In contrast, both DIO1 expression and intracellular T3 concentrations progressively decrease in MASH. 70 These findings suggest that there is an “intrahepatic hypothyroidism” phenotype in MASH that decreases lipophagy and β-oxidation of fatty acids, leading to overaccumulation of saturated long-chain free fatty acids (FFAs) and lipotoxicity in the liver. 43 TH treatment of mice with MASH restores lipophagy, β-oxidation of fatty acids, and mitochondrial activity to reduce inflammation and fibrosis. Increased DIO3 expression may also occur in MASH to further reduce intrahepatic T3 concentration. 71 Additionally, decreased expression of THRβ in the liver occurs in MASH, further impairing TH signaling. 72 Interestingly, epigenetic regulation of DIO1 via the induction of miR-34a-5p and THRβ by DNA methylation of the first intron of intron 1 of the THRB gene is seen in liver tissue obtained from patients with MASH. 73
TH and thyromimetic therapy for MASLD
The rising prevalence of MASLD/MASH in combination with the paucity of U.S. Food and Drug Administration (FDA)-approved pharmacological agents for the treatment of MASH has been a major challenge in the management of the disease. Numerous epidemiological studies show that patients with hypothyroidism and subclinical hypothyroidism have a higher risk for MASLD, and the severity of the disease correlates with thyroid dysfunction. 45 –51 Interestingly, transcriptome analyses of liver tissue from patients with obesity undergoing bariatric surgery reveal that the top-ranking altered gene set comprises genes that are positively regulated by TH. 74 A clinical study of diabetic patients with MASLD shows significant improvements in hepatosteatosis when they are treated with low doses of LT4. 70 Studies in patients with RTHβ who harbor a dominant negative mutant THRβ have increased hepatic fat content and higher risk for fibrosis than their sibling controls with wild-type (WT) THRβ alleles, suggesting there is an important role for THRβ in the pathogenesis of MASH. 75 These clinical and earlier preclinical studies showing TH increasing autophagy and β-oxidation to reduce hepatosteatosis and improve inflammation and fibrosis in MASH helped to establish the theoretical foundation and rationale for the development of thyromimetics as a therapy for MASH. 55,61
Several thyromimetic compounds with THRβ selectivity, such as Sobetirome and Eprotirome, showed early promise as therapies for hypercholesterolemia ∼20 years ago. However, their development was halted due to bone- and cartilage-associated side effects and the availability of multiple drugs for the treatment of hypercholesterolemia that were already available in the market. 76,77 Similarly, TERN-501, another oral THRβ agonist developed to treat hypercholesterolemia and MASH, demonstrated significant reduction in liver fat content (via magnetic resonance imaging-derived proton density fat fraction [MRI-PDFF]) and inflammation (via magnetic resonance imaging iron-corrected T1 mapping) but was not developed further. 76 In contrast, Resmetirom, formerly known as MGL-3196, is an orally administered liver- and THRβ-selective drug that recently was shown to be effective for the treatment of adult patients with noncirrhotic MASH and received conditional approval by the FDA as therapy in conjunction with diet and exercise. 78 This landmark approval is the culmination of the preclinical and clinical work, demonstrating the efficacy of TH for the treatment of MASLD and MASH. 2 Due to its tissue- and receptor isoform-selectivity, Resmetirom harnesses the beneficial effects of T3 in the liver while also mitigating the cardiovascular and bone side effects due to TH activation of THRα, the THR isoform, which is highly expressed in those tissues. Currently, Resmetirom is also under regulatory review in the European Union. 79
In mouse models of MASH with fibrosis, Resmetirom improves hepatic metabolic profiles, fibrosis, and non-alcoholic fatty liver disease (NAFLD) activity score. 80 In a phase 2 randomized placebo-controlled trial involving 125 subjects with noncirrhotic MASH, 80 mg per day Resmetirom significantly reduces liver fat content (via MRI-PDFF) by 28.4% over 36 weeks. Liver enzymes and fibrosis biomarkers (such as Enhanced Liver Fibrosis (ELF) and pro-peptide of type III collagen) also improve, as well as health-related quality of life indices. 58,81 The phase 3 MAESTRO-NAFLD-1 trial confirms the acceptable safety profile of Resmetirom 80 or 100 mg/day in 1143 obese subjects with MASH over 52 weeks. Resmetirom was well tolerated, with only transient and mild–moderate diarrhea and nausea occurring more frequently in treated patients than in patients taking a placebo. Importantly, there are no disturbances of endocrine function reported. 82 In the phase 3 MAESTRO-NASH trial, 966 obese adults with biopsy-proven noncirrhotic MASH are given 80 mg and 100 mg doses of Resmetirom versus placebo over 52 weeks. Both doses of Resmetirom are superior to placebo in terms of MASH resolution without any worsening of fibrosis (25.9–29.9% vs. 9.7%, p < 0.001 for both comparisons with placebo) and the reduction of fibrosis by at least one stage (24.2–25.9% vs. 14.2%, p < 0.001 for both comparisons with placebo). There are also improvements in other surrogate markers such as MRI-PDFF, liver enzyme levels, liver stiffness scores, and ELF measured by Fibro scanning. However, some overweight patients treated with 80 mg have a suboptimal response. Therefore, the FDA recommends Resmetirom be administered at 100 mg per day in patients who weigh more than 100 kg. 83 Resmetirom also does not have significant effects on body weight, blood glucose levels, and insulin resistance 84 ; however, it lowers serum lipid levels by 13.6–16.3% for LDL-C, 21.7–22.7% for TG, and 30.4–35.9% for Lp(a) levels. 83 In a separate 12-week phase 2 randomized placebo-controlled trial investigating 116 subjects with heterozygous familial hypercholesterolemia who did not reach guideline-recommended LDL-C levels with standard therapy, Resmetirom further lowers LDL-C levels by 18.8% compared with placebo (p < 0.0001) with a mean reduction of 27 mg/dL (p < 0.0001). TG, apoB, and Lp(a) levels are also significantly reduced in these patients. 85
Another THRβ-selective agonist, VK2809, is currently in late-stage clinical development for the treatment of MASH. The results on the phase 2b VOYAGE clinical trial of VK2809 in MASH were recently presented at the annual meeting of the American Association for the Study of Liver Disease in November 2024. 86 VK2809 reduces hepatic fat content by 37–55% compared with baseline after 52 weeks of treatment. VK2809 significantly improves fibrosis (51% vs. 34%, p = 0.03), lipid profiles (LDL-C, TGs and TC, and TC), and MASH resolution (69% vs. 29%, p < 0.0001).
Taken together, these data are encouraging and could herald a new “dawn” for thyromimetics, especially for metabolic disorders in which affected tissues may have low intracellular T3 concentrations despite having normal serum TH levels. More information is needed about strategies for monitoring clinical response, employing combination therapy, and observing long-term effects of these agents for both clinical outcomes and safety. Given the efficacy of low-dose LT4 for hepatosteatosis in patients with diabetes and TH therapy for MASH in mice, 70,87 it is reasonable to speculate that low-dose T3 or T4 could be alternative treatments for hepatosteatosis and/or MASH. If so, low-dose TH treatment for MASH will be a safe and cost-saving alternative to thyromimetics, particularly for patients from low-income countries. Further clinical studies are needed to confirm the efficacy and safety of low-dose TH therapy in patients with MASH. GLP-1 receptor agonists, although not yet approved specifically for MASLD, have been shown to reduce liver fat, improve steatohepatitis, and slow fibrosis progression in patients with T2DM and obesity. 88 Unlike TH analogs that mainly target the liver, GLP-1 receptor agonists improve the broader metabolic problems seen in MASLD, including weight gain, hyperglycemia, and cardiovascular disease. Their wide metabolic benefits and high safety profile make them a promising treatment option for MetS-related conditions.
Nonthyroidal illness syndrome effects on the liver
Nonthyroidal illness syndrome (NTIS) or sick euthyroid syndrome is characterized by an ensemble of changes in serum TH concentrations that typically occur during severe illness. NTIS is typically characterized by a low T3, normal or reduced T4, increased rT3, and low, normal, or slightly elevated TSH levels. 89 These changes in TH levels correlate with morbidity and mortality. The pathophysiology of NTIS involves several organs, including the liver, and features changes in many aspects of TH signaling, such as the expression of circulating transport proteins, TH transporters, DIOs, and THR activity.
TH transport to the human liver is affected by chronic illness owing to decreased hepatic production of circulating TH-binding proteins such as TBG, TTR, and HSA to decrease total serum T4 levels. Circulating TH-binding proteins also exhibit changes in their binding affinity during NTIS to alter free and total TH levels in patients’ sera during acute and prolonged illness. 90 Accumulation of bilirubin (BIL) and nonesterified fatty acids in the liver reduces T4 uptake and T4 to T3 conversion in NTIS. 91 Interestingly, MCT8 expression is increased during acute illness but is suppressed during prolonged critical illness. 92 Therefore, the initial upregulation of MCT8 expression may represent a compensatory mechanism to increase intrahepatic T3 levels when circulating T3 concentrations are low. 93 On the contrary, the suppression of MCT8 expression during chronic illness may contribute to the worse prognosis of patients with “low T3” syndrome. Chronic illness also markedly downregulates the expression of DIO1. However, DIO3 expression may be differentially regulated depending on the duration of the illness. DIO3 expression decreases during acute illness and increases during prolonged illness, most likely due to changes in energy requirements in the liver during these phases of illness. 94,95
Hepatic THRα and THRβ mRNA expression are differentially affected depending on the severity and duration of the illness. Acute illness reduces hepatic THRα and THRβ expression in rodents but has no effect on their expression in chronic inflammation and sepsis. 96,97 On the contrary, biopsies prior to transplantation in patients with chronic liver disease show increased THR expression associated with low serum T3 and T4 levels. 98 However, mRNA expression of hepatic THRα1, THRα2, and THRβ1 and TH-regulated target genes are unchanged despite altered serum TH levels. Taken together, these findings suggest that despite the changes affecting circulating TH levels, the liver employs various compensatory measures to regulate DIO1, DIO3, and THR expression in its attempt to maintain an “euthyroid-like” status in the liver. 99
TSH regulation of liver function
Thyrotropin receptors (TSHRs) are Gs-coupled proteins located on the cell membranes of thyrocytes that normally bind to TSH. However, in Graves’ disease (GD), autoimmune antibodies (TRAbs) bind to TSHRs to stimulate TH synthesis and secretion and cause hyperthyroidism. In addition to their expression in the thyroid gland, TSHR expression in nonthyroid cells, such as retro-orbital fibroblasts and their activation by TRAbs, plays a pivotal role in the pathogenesis of Graves’ orbitopathy. Interestingly, Zhang et al. also demonstrated the presence of functional TSHR protein in human hepatocytes, suggesting the possibility that TSH may bind to TSHRs in the liver to induce multiple effects on lipid metabolism. 100 In experimental models, TSH has been shown to increase hepatic TG content via upregulation of sterol regulatory element-binding proteins (SREBP-1c) activity, repress bile acid synthesis through a SREBP-2/HNF-4α/CYP7A1 pathway, and stimulate HMG-CoA reductase activity by reducing its phosphorylation through inhibition of AMP-activated protein kinase. 49,101,102 TSH has also been reported to activate hepatic gluconeogenesis by promoting CRTC2 activation via the TSHR/cAMP/PKA pathway. 103 However, these findings are based largely on preclinical studies, and the physiological relevance of hepatic TSH signaling in humans remains to be fully established. Further studies are needed to clarify the extent to which TSH exerts meaningful metabolic effects on the liver, particularly in clinical conditions such as hypothyroidism or GD.
Finally, hypothyroidism has effects on protein metabolism. There is reduced gluconeogenesis and anapleurosis into the tricarboxylic acid (TCA) cycle, leading to lower protein catabolism. Serum AST and LDH levels can increase during hypothyroidism, and their elevation may be partly due to myopathy induced by low circulating TH levels. 23,24
Resistance to TH and the liver
Mutations in THRα and THRβ cause two different syndromes of resistance to TH (RTHα and RTHβ) with distinct phenotypes. The clinical features of these two types of RTH have been described elsewhere. 104,105 Generally, the mutant THRs have dominant negative activity over the other THRs expressed in the cells if the mutant THR is highly expressed in that cell type. Of note, most patients with RTHβ have elevated serum TH levels with nonsuppressed TSH levels due to pituitary resistance to circulating TH levels, whereas patients with RTHα have slightly reduced T4, high T3, and normal TSH levels. RTHα patients have bradycardia, constipation, anemia, and growth retardation due to expression of high levels of mutant THRα in the heart, gut, erythrocyte precursors, and bone. RTHβ patients exhibit decreased and normal function in many tissues that express mutant THRβ despite having high circulating TH levels. However, they also have “hyperthyroid-like” symptoms in the heart and bone since these tissues express high amounts of THRα relative to THRβ.
THRα is highly expressed in intestinal epithelial cells, so its mutation causes impaired differentiation and maturation of enterocytes, leading to abnormal intestinal structure and function. Accordingly, patients with RTHα may have reduced nutrient absorption and constipation due to altered gut motility even though liver function is not significantly affected. Patients with RTHβ have more severe metabolic disturbances in the liver than those with RTHα, since THRβ is the predominant THR isoform expressed in hepatocytes. In RTHβ, the mutant THRβ has impaired TH-binding affinity and dominant negative activity on WT THRs, resulting in decreased expression of important metabolic genes involved in lipid (e.g., CPT1a, LCAD) and cholesterol (e.g., LDL receptor [LDLr], HMG-CoA reductase) homeostasis. 106 Additionally, TH plays a key role in hepatic lipophagy and mobilization of TGs from fat droplets to lysosomes for hydrolysis into glycerol and free fatty acids. Therefore, patients with mutations in THRβ may also have impaired hepatic lipophagy, contributing to decreased β-oxidation of fatty acids that is observed in preclinical models of MASH. 107 Indeed, RTHβ can increase serum cholesterol and TG levels by mimicking features of hepatic metabolism that resemble those seen in the “intrahepatic hypothyroidism” of MASH. Accordingly, hepatic lipophagy and β-oxidation are likely impaired in RTHβ patients, leading to hepatic lipid accumulation and MASLD. Indeed, in a large family of RTHβ patients, there is a higher prevalence of hepatosteatosis and elevated risk for fibrosis than their sibling controls. 75 These studies also demonstrate the critical role of THRβ in the liver to maintain normal lipid metabolism and prevent the development of MASLD in humans. Additionally, since insufficient TH action decreases gluconeogenesis, hepatic glucose production may be impaired during fasting in some RTHβ patients. Table 2 compares the gastrointestinal and liver effects of RTHα with RTHβ.
Comparison of Resistance to Thyroid Hormone Alpha (RTHα) and Resistance to Thyroid Hormone Beta (RTHβ) in Gastrointestinal and Liver Effects
fT3, free triiodothyronine; fT4, free thyroxine; GI, gastrointestinal; RTH, resistance to thyroid hormone; TT3, total triiodothyronine; TT4, total thyroxine.
Hyperthyroidism Effects on Liver Function
Causes of hyperthyroidism
Thyrotoxicosis is caused by increased synthesis and secretion of TH by the thyroid (e.g., GD, toxic nodular goitre); excessive release of stored preformed TH from the thyroid gland (e.g., acute thyroiditis); or extrathyroidal and exogenous sources of THs. 108 The most common cause of hyperthyroidism is GD, which has a global prevalence of 2% in women and 0.5% in men. 109 GD is an autoimmune disease in which autoantibodies directed against the thyroidal TSH receptor (TSHR) cause increased TH synthesis and secretion. During hyperthyroidism, increases in TH levels accelerate the metabolism in the liver, leading to stimulation of hepatic glucose production and fatty acid and cholesterol metabolism. Hyperthyroidism induces the expression of numerous hepatic enzymes and may cause hepatic dysfunction in severe cases.
Hyperthyroidism effects on hepatic metabolism
The levels of TC, LDL-C, ApoB, and Lp(a) tend to decrease in patients with clinical or subclinical hyperthyroidism. This decline is due to increased LDLr gene expression, resulting in enhanced LDLr-mediated catabolism of LDL particles. 110,111 Hyperthyroidism induces LDL oxidation that is dependent upon serum fT4 levels. 112 Hyperthyroidism decreases HDL-C levels in hyperthyroidism, due to increased CETP-mediated transfer of cholesteryl esters from HDL to VLDL and increased hepatic lipase-mediated catabolism of HDL2 110,111 TG levels usually remain unchanged in hyperthyroidism. Treatment of hyperthyroidism with ATDs, RAI, or surgery reverses these abnormalities in lipid metabolism although the minor changes in subclinical hyperthyroidism can be refractory to treatment. 35
Hyperthyroidism is associated with elevated endogenous hepatic glucose production due to increased gluconeogenesis and glycogenolysis, enhanced glucose absorption from the gastrointestinal tract, and development of hepatic insulin resistance, all of which contribute to glucose intolerance. 113 Excess TH also accelerates insulin degradation and decreases the half-life of insulin. 114 The induction of hepatic glucose output due to gluconeogenesis and glycogenolysis also causes hyperinsulinemia, glucose intolerance, and promotes peripheral insulin resistance. The development of thyrotoxicosis worsens preexisting T2DM and may induce diabetic ketoacidosis since increased lipolysis in adipose tissue and increased hepatic β-oxidation increase ketone body production. 115,116
Serum SHBG and TH (fT3 and fT4) levels are positively correlated with each other. SHBG levels are increased in hyperthyroidism and normalize when thyroid status is restored after treatment. Clinically, measurement of serum SHBG has been used to distinguish TSH-secreting pituitary adenoma and patients with RTHβ syndrome in patients with high circulating TH levels. Hyperthyroid patients can also present with increased serum ferritin concentrations that resolve after treatment. However, as mentioned above, the serum levels of SHBG and ferritin concentrations may be altered by external factors and thus may not be reliable indicators of intrahepatic TH status. Moreover, although serum SHBG has been proposed as a measurable indicator of intrahepatic TH levels, its discriminatory power is limited. Other potential liver tissue biomarkers that reflect hyperthyroid status are glycerophosphate dehydrogenases (G6PH) and CD5L. Although G6PH may be useful in assessing intrahepatic TH status, 117 it cannot be measured in the serum, thus limiting its utility. CD5L is a protein involved in the modulation of leukocyte function with potential as a marker for hepatic TH status since its levels correlate well with serum fT3 concentrations. 118 However, the influence of inflammatory processes, age, gender, and various diseases on the latter’s serum levels has not been carefully examined.
Hyperthyroidism and liver function abnormalities
Liver function test (LFT) abnormalities commonly occur in patients with newly diagnosed hyperthyroidism. In untreated hyperthyroidism, one in two patients has LFT abnormalities (e.g., alkaline phosphatase [ALP], ALT, AST, GGT, and BIL) irrespective of ALP elevations. The prevalence of LFT abnormalities is higher in GD patients, occurring in two-thirds of this subgroup. 119 The degree of LFT elevation ranges from borderline (<2 × ULN) to mild elevation (2–5 × ULN), although there is wide variation among affected patients. Several mechanisms may account for the LFT changes found in hyperthyroidism, including direct liver toxicity from prolonged exposure to excessive TH, leading to increased mitochondrial activity and ROS to cause cell damage; hepatocyte damage from excessive gluconeogenesis and protein degradation; and concomitant congestive hepatopathy (necrosis) due to thyrotoxic heart failure. 120 These hepatocellular injuries and hemodynamic changes are exacerbated in thyroid storm, which in combination with higher oxygen demand and ROS damage by mitochondria can cause severe hepatitis and liver failure. 121,122 In GD, TSH receptor antibodies (TRAb) may also cause additional inflammatory injury to hepatocytes. 123 Initial serum TSH concentrations <0.02 mIU/L, male gender, and African American race are significant predictors of abnormal serum LFTs 6 months after the diagnosis of new-onset thyrotoxicosis. 124 In a meta-analysis to assess the prevalence and the response to anti-thyroid drug (ATD) therapy of LFT abnormalities in newly diagnosed and uncomplicated hyperthyroidism, ALP elevation is the most common LFT abnormality, followed by ALT, AST, GGT, and BIL. 119 ALP also originates from the bone due to increased osteoblast enzymatic activity, so it needs to be interpreted with caution. ALT and AST are released into the circulation whenever there is hepatocyte injury and/or cell death (hepatitis), so they are more reliable liver-specific markers. Since GGT is abundant in the liver but not in bone, its measurement is able to confirm whether an elevated ALP originates from the liver. BIL elevation is rare and may be due to hepatobiliary dysfunction rather than liver injury. Of note, TH excess has increased prevalence of cholestasis, as many hyperthyroid patients display increases in serum levels of ALP, GGT, and BIL and have histological evidence of centrilobular intrahepatic cholestasis. 125
The mainstay of medical management of GD is the thioamide compounds, methimazole (MMI), its prodrug derivative, carbimazole (CMZ), and PTU. The presence of LFT abnormalities in newly diagnosed patients with hyperthyroidism (mainly GD) presents a clinical conundrum to clinicians that may cause them to be hesitant about initiating antithyroid drug treatments. Indeed, thioamides are associated with rare drug-induced liver injury (DILI), with an overall incidence of 0.03–0.07% that most often occurs in the first 3 months of therapy. 126,127 CMZ/MMI-induced hepatotoxicity is often cholestatic, whereas PTU is associated with hepatocellular damage, although recent reports from Asia suggest ethnicity and genetic factors may modify their presentation as described in the literature. 128,129 PTU is the third most common cause of DILI in patients under the age of 20 years old. Studies indicate that ∼10% of patients who develop severe liver damage from PTU may progress to liver failure and require liver transplants. 130 However, given the relatively low risk for initiating or exacerbating liver disease, ATDs are safe for most patients with hyperthyroidism. Several clinical studies have evaluated the changes in LFT abnormalities with ATD treatment. In most cases, ATDs normalized LFTs within 12 months of treatment in patients with elevated serum AST, ALT, and GGT levels. Although transient elevations of ALT have been observed 6–8 weeks after starting therapy, it is usually resolved after 3 months of treatment. 131 –133 The isolated rise in serum ALP observed after the treatment of GD is related to heightened osteoblastic activity during ATD treatment. 134 –136 Taken together, these data suggest that abnormal LFTs are not an absolute contraindication to ATD therapy, although diligent monitoring of liver status and careful assessment of underlying liver disease need to be considered if baseline transaminases are more than five times the upper limit of the normal range. 129
Iatrogenic effects on the liver due to treatment of thyroid disease
Treatment of hypothyroidism with LT4 is usually not associated with liver function abnormalities if administered at proper doses. However, there are a few cases in the literature in which patients develop liver dysfunction after taking replacement doses of LT4. It is likely that some of these patients have a genetic predisposition and/or idiosyncratic response to the drug formulation. In some cases, liver dysfunction is reversed when therapy is switched from LT4 to T3, suggesting that excessive DIO activity may be contributory. 137 On the contrary, overtreatment of hypothyroidism leads to iatrogenic thyrotoxicosis and can cause liver dysfunction similar to that seen in endogenous hyperthyroidism. 120
Liver dysfunction due to hyperthyroidism occurs for several reasons outlined earlier (in the sections “Hyperthyroidism Effects on Hepatic Metabolism” and “Hyperthyroidism and Liver Function Abnormalities”). Although a direct correlation between severity of changes in LFTs with serum TH levels has not been established for hyperthyroidism, liver dysfunction usually improves when hyperthyroidism resolves after initiation of first-line thioamide treatment. However, in patients with severe cases of liver dysfunction and refractory hyperthyroidism, thioamides should be withdrawn and be replaced by early definitive treatment with RAI or surgery.
Interestingly, several studies have investigated the impact of liver dysfunction on the outcome of RAI treatment of GD. In a study of 2385 patients with GD undergoing RAI treatment, various degrees of liver function abnormalities were noted in 65% of the cases. 138 Liver dysfunction normalizes in 77.3% of these patients within 6 months after RAI treatment. The prognosis of hepatic dysfunction is also associated with the outcomes of RAI treatment of GD in this study. The remission rate of hepatic dysfunction in the cured group of patients (defined as those achieving hypothyroidism or euthyroidism) is higher (90.8%) than in the group of patients with persistent hyperthyroidism (41.8%). Unfortunately, these results are not replicated in a more recent study involving 510 consecutive patients with GD, in which 222 have hepatic dysfunction after receiving their first RAI treatment. However, the percentage of achieving euthyroidism or hypothyroidism is higher in patients with liver dysfunction than in those with normal liver function 3 months (74.5% vs. 62.5%, p = 0.005) and 6 months (82.1% vs. 69.1%, p = 0.002) after RAI treatment, but not significant after 12-month follow-up (89.6% vs. 83.2%, p = 0.081). No significant differences in treatment efficacy, recurrence rate, or incidence of early onset hypothyroidism are observed between the groups with or without hepatic dysfunction. 139
Thyroid storm is an uncommon but life-threatening endocrine emergency characterized by extreme manifestations of thyrotoxicosis. Hepatic injury as part of the multi-organ dysfunction due to thyroid storm ranges from deranged liver enzymes to acute liver failure and fulminant hepatitis. In severe liver failure, the use of thioamides may risk further aggravation and worsen liver injury, necessitating their discontinuation. Hence, several authors have reported successful utilization of therapeutic plasma exchange (TPE) as a bridge to urgent thyroidectomy in such circumstances. 140,141 TPE has a Class II indication for thyroid storm in the 2023 American Society for Apheresis guidelines, either as a stand-alone or adjunct therapy. 142 TPE is usually performed in patients with thyroid storm with severe symptoms and when the patient does not improve with first-line therapies within 24–48 hours of treatment or when first-line therapies cannot be used due to toxicity. Apart from TPE, the use of molecular adsorbent recirculating system with or without RAI has been reported as an effective treatment for thyroid storm with severe liver injury. 143
Persistent changes in the liver after correction of hyperthyroidism
Most patients with abnormal serum LFTs and hyperthyroidism normalize their LFTs after ATD treatment, and their serum TH levels return back to baseline. 119 However, some patients can have residual nonspecific symptoms such as neuropsychiatric complaints, fatigue, and social isolation 144,145 despite normalization of serum TH and TSH levels. Although serum cholesterol and lipid profiles can increase after correction to euthyroidism, 146 there has not been further study of the persistent long-term metabolic changes in the liver after correction of hyperthyroidism in man. In a mouse model of hyperthyroidism, long-term changes in hepatic gene expression and metabolism persist after serum TH and TSH levels are normalized. 147,148 The mechanism for these long-term effects is not known but may be due to residual epigenetic changes induced by chronic hyperthyroidism. TH increases hepatic cytochrome P450 (CYP), which is important for drug metabolism and detoxification. 149 In this connection, it is important to observe and monitor the pharmacokinetics of other drugs taken by the patient during the transition from hyperthyroidism to euthyroidism and adjust their doses accordingly.
Effects of Liver Disease on the Thyroid
Liver diseases affecting the thyroid
Liver diseases and their treatment can affect thyroid function as reflected by changes in TH indices on laboratory testing. 150,151 Although patients generally remain clinically euthyroid in acute hepatitis and cirrhosis, thyroid function can be affected by specific liver diseases or treatments of these conditions. In many cases, breakdown of immune tolerance with the triggering of autoimmunity appears to be the underlying cause. For example, patients with autoimmune liver diseases (primary biliary cholangitis [PBC], autoimmune hepatitis [AIH], and primary sclerosing cholangitis [PSC]) are at higher risk for developing autoimmune thyroid disease. 152 –154 Cross-reactivity by the antithyroid and antiliver autoantibodies for similar or the same cell surface antigens in both tissues, and the recruitment of autoreactive T cells may underlie this frequent association. Moreover, Hashimoto’s thyroiditis occurs more often in PBC and AIH, whereas GD is more commonly found in PSC. However, the presence of thyroid disease does not seem to influence the course or prognosis of the underlying liver condition. Although both hepatitis C infection and its treatment with interferon alpha or ribavirin have been associated with thyroid function abnormalities, newer treatments with direct-acting antivirals do not affect thyroid function. Potential alterations in thyroid function associated with liver diseases and their treatments are summarized in Table 3.
Potential Alterations in Thyroid Function Associated with Liver Diseases and Their Treatments
Conclusion
THs play a key role in the homeostatic regulation of hepatic lipid, carbohydrate, and protein metabolism. Therefore, both hypothyroidism (frequently in the form of Hashimoto’s thyroiditis) and hyperthyroidism (usually GD, toxic multinodular goitre, and iatrogenic causes) can have profound effects on hepatic metabolism, leading to systemic effects since hypothyroidism increases circulating LDL-C and TG levels, decreases body energy expenditure by reducing basal metabolic rate and thermogenesis, and increases the risks for diabetes, obesity, MetS, and MASLD. On the contrary, hyperthyroidism frequently affects LFTs and, in severe cases, can lead to hepatitis and liver damage due to excessive mitochondrial activity and generation of ROS that damage cellular proteins. By and large, the systemic effects of hyperthyroidism on hepatic cholesterol and fatty acid metabolism are the opposite of those observed in hypothyroidism. Given the many effects of the thyroid and THs on metabolism and energy utilization, and the important roles of the liver in these processes, clinicians need to recognize their reciprocal effects on each other when there are pathological conditions affecting one or both organs.
Since MASLD and MetS are frequently associated with hypothyroidism, it is recommended that patients with these conditions be screened for thyroid dysfunction. Likewise, patients with hypothyroidism should be evaluated for dyslipidemia, MetS (including hypertension, cardiovascular disease, and diabetes), and MASLD. Patients with elevated LFTs and no underlying liver disease should be evaluated for hyperthyroidism. Additionally, autoimmune liver disease is associated with a higher risk for autoimmune thyroid disease and vice versa, so they need to be considered when either is encountered in the patient.
Currently, it is difficult to assess intrahepatic TH concentration, which appears to be diminished in MASLD due to downregulation of DIO1. Determining intrahepatic concentrations before and after treatment would be clinically important to determine which patients are responding to thyromimetic therapy for MASH. Rodent studies suggest there is an initial compensatory increase in DIO1 activity during steatosis and downregulation of DIO1 activity as MASH progresses. 43 SHBG and ferritin are potential serum markers for disease progression and therapeutic response to thyromimetics, but their utility needs to be fully evaluated over a range of TH levels in larger populations of patients. Metabolomic profiling offers the possibility of identifying novel TH-regulated serum metabolites or profiles of serum metabolites that are generated specifically by the liver and enable assessment of intrahepatic TH status.
Resmetirom was recently approved by the FDA as the first pharmacological treatment for MASH. Other liver- or THRβ-selective thyromimetics are currently being developed for the treatment of MASH. It is also possible that Resmetirom or other thyromimetics could be administered with other new drugs to treat MASH, such as analogs and GLP-1 receptor agonists, to increase their efficacy in reducing steatosis, inflammation, and fibrosis in MASH. It remains to be seen whether low-dose T4 or T3 could have similar beneficial effects on MASH. However, given the continuous, J-shaped association between fT4 and cardiovascular and all-cause mortality (even within the reference range), the safety of low-dose LT4 therapy for MASLD cannot be assumed and would need to be carefully titrated. 155 These findings underscore the need for prospective trials to evaluate both the efficacy of treatment and potential risks, particularly in older individuals. If T3 or T4 are effective, they certainly would help patients in developing countries who otherwise would not be able to afford thyromimetics or other new pharmacological therapies for MASH. The possibility that thyromimetics and TH can be used to treat MASH is an exciting prospect and raises the possibility that TH may also be able to treat extrahepatic manifestations of MetS that are associated with MASLD.
Footnotes
Authors’ Contributions
C.-L.C.: Conceptualization (supporting), writing—original draft (lead), and writing—review and editing (equal). G.B.B.G.: Writing—original draft (supporting) and writing—review and editing (supporting). P.M.Y.: Conceptualization (lead), writing—original draft (supporting), and writing—review and editing (equal). All the authors have approved the final article for publication and are accountable for this work.
Author Disclosure Statement
The authors have no relevant disclosures to report.
Funding Information
No funding was received for this review.