
Abstract
Background:
DICER1 plays a central role in microRNA biogenesis and functions as a tumor suppressor in thyroid cancer, which is the most frequent endocrine malignancy with a rapidly increasing incidence. Thyroid cancer progression is associated with loss of cell differentiation and reduced expression of thyroid differentiation genes and response to thyrotropin (TSH). Here we investigated whether a molecular link exists between DICER1 and thyroid differentiation pathways.
Methods:
We used bioinformatic tools to search for transcription factor binding sites in the DICER1 promoter. DICER1, NKX2-1, PAX8, and CREB expression levels were evaluated by gene and protein expression in vitro and by interrogation of The Cancer Genome Atlas (TCGA) thyroid cancer data. Transcription factor binding and activity were assayed by chromatin immunoprecipitation, band-shift analysis, and promoter–reporter gene activity. Gene-silencing and overexpression approaches were used to elucidate the functional link between DICER1 and differentiation.
Results:
We identified binding sites for NKX2-1 and CREB within the DICER1 promoter and found that both transcription factors are functional in thyroid cells. TSH induced DICER1 expression in differentiated thyroid cells, at least in part, through the cAMP/PKA/CREB pathway. TCGA analysis revealed a significant positive correlation between CREB and DICER1 expression in human thyroid tumors. NKX2-1 overexpression increased DICER1 promoter activity and expression in vitro, and this was significantly greater in the presence of CREB and/or PAX8. Gain- and loss-of-function assays revealed that DICER1 regulates NKX2-1 expression in thyroid tumor cells and vice versa, thus establishing a positive feedback loop between both proteins. We also found a positive correlation between NKX2-1 and DICER1 expression in human thyroid tumors. DICER1 silencing decreased PAX8 expression and, importantly, the expression and activity of the sodium iodide symporter, which is essential for the diagnostic and therapeutic use of radioiodine in thyroid cancer.
Conclusions:
The differentiation transcription factors NKX2.1, PAX8, and CREB act in a positive feedback loop with DICER1. As the expression of these transcription factors is markedly diminished in thyroid cancer, our findings suggest that DICER1 downregulation in this cancer is mediated, at least partly, through impairment of its transcription.
Introduction
The highly conserved RNAse III enzyme DICER1 is crucial for the biogenesis of microRNAs (miRNAs) by cleaving double-stranded small-noncoding RNA precursors to generate mature miRNAs of 21–24 nucleotides. Several studies have highlighted an association between the reduced expression of DICER1 and poor prognosis in different cancers, including lung, breast, skin, endometrial, and ovarian cancer (1 –5). We recently showed that DICER1 acts as a tumor suppressor in thyroid cancer, as its downregulation promotes proliferation, migration, and invasion (4). We also found that low DICER1 levels in thyroid carcinomas correlate with a worse clinical outcome (3).
Thyroid cancer is the most frequent endocrine malignancy whose incidence has increased (6). Well-differentiated papillary thyroid cancer (PTC) and follicular thyroid cancer (FTC) account for most cases and usually have a good prognosis. By contrast, undifferentiated anaplastic thyroid cancer (ATC) is an extremely aggressive cancer with a five-year disease-free survival rate of almost 0%. The classical view of thyroid cancer pathogenesis considers thyroid carcinomas as tumors that progress through a dedifferentiation process, which underscores the need to identify therapeutic targets that drive thyroid cancer differentiation (7).
Thyroid differentiation is finely tuned by positive and negative regulatory signals (8,9). Thyrotropin (TSH) engages its cognate G protein-coupled receptor, TSH receptor (TSHR), to activate cAMP/PKA/CREB signaling, which regulates the transcription of thyroid-specific genes such as thyroglobulin, thyroperoxidase, and sodium iodide symporter (NIS) (10,11). In turn, these genes are regulated by the instructive transcription factors NKX2-1, PAX8, and FOXE1, which define the differentiated thyroid phenotype (12,13).
TSHR signaling is required for thyroid carcinogenesis in a mouse model of spontaneous FTC (14). In addition, dedifferentiated thyroid tumors lose their response to TSH and its downstream signaling (15,16). Importantly, thyroid cancer shows a marked downregulation of differentiation, and consequently, a characteristic decrease in the expression of thyroid transcription factors is critical for cancer progression (11,13).
Normal developmental processes and cancer share many pathways related to cell proliferation and differentiation. In this respect, conditional deletion of Dicer1 in thyroid follicular cells during mouse development causes a strong reduction in the expression of differentiation markers such as Nkx2-1 and Pax8 (17) and NIS later in adults (18). Moreover, mice are hypothyroid at birth and present characteristics of neoplastic alterations in thyroid tissue in adult life (17). These findings suggest that miRNAs are necessary to maintain thyroid tissue homeostasis.
Given the crucial role of DICER1 in thyroid cancer aggressiveness, the present study was designed to uncover the transcriptional mechanisms that control its expression. We also explored whether DICER1 downregulation is involved in differentiation by regulating the instructional transcription factors involved in this process.
Methods
Bioinformatic predictions
Transcription factor binding sites within the human DICER1 promoter were identified using the ECR browser. The cBioPortal for Cancer Genomics was used to obtain the correlations between DICER1, CREB, and CREM mRNA levels, on the thyroid carcinoma data set of The Cancer Genome Atlas (TCGA) database.
Patients
Paired samples of PTC tumors and contralateral normal thyroid tissue from patients (n = 7) were studied as previously described (19). Written informed consent was obtained from all the patients following the protocols approved by the Hospital ethics committee.
Cell culture and transfection
Cells, culture methods, and transfection (20) are described in Supplementary Data.
Luciferase assay
The pGL3-DICER-Prom vector or serial deletion mutants were transfected together with pCMV-Renilla and the corresponding transcription factor expression vectors. Promoter activity was assayed as described previously (9).
RNA quantification, protein extraction, and Western blotting
RNA quantification, protein extraction methods, and Western blotting are described in Supplementary Data. The primers used are described in Supplementary Table S1.
DNA-NKX2.1 binding assays
Chromatin immunoprecipitation (ChIP) and electrophoretic mobility shift assay (EMSA) details are described in Supplementary Data.
Iodide transport
PCCl3 cells were assayed for iodide transport as previously described (21), and are detailed in the Supplementary Data.
Statistical analysis
Results are expressed as the mean ± standard deviation of at least three different experiments performed in triplicate. Statistical significance was determined by Student's t-test (two-tailed) analysis and differences were considered significant at a p-value <0.05.
Results
DICER1 promoter contains CRE motifs and binding sites for NKX2-1
DICER1 acts as a novel tumor suppressor and is downregulated in thyroid cancer (3); however, the mechanism of this downregulation and its transcriptional regulation in the thyroid gland is unknown. We conducted in silico analysis of consensus transcription factor binding sites in the human DICER1 promoter, considering 2500 base pairs upstream of the transcription initiation site. We identified two canonical CRE motifs at positions −1876 (consensus sequence AGTCAt) and −1856 (consensus sequence TGTCAt) and 10 NKX2-1 consensus binding sites (CAAG) at −102, −308, −696, −1159, −1512, −1528, −1751, −2109, −2138, and −2224 (Fig. 1). Of note, CREB, the main transcription factor that binds to CRE elements, and NKX2-1 have fundamental roles in thyroid differentiation (13), and their loss is a hallmark of thyroid cancer progression. Accordingly, the decrease in DICER1 levels in patients with thyroid cancer could be explained by the dedifferentiation events occurring in tumors. For these reasons, we examined the roles of these transcription factors in DICER1 regulation.
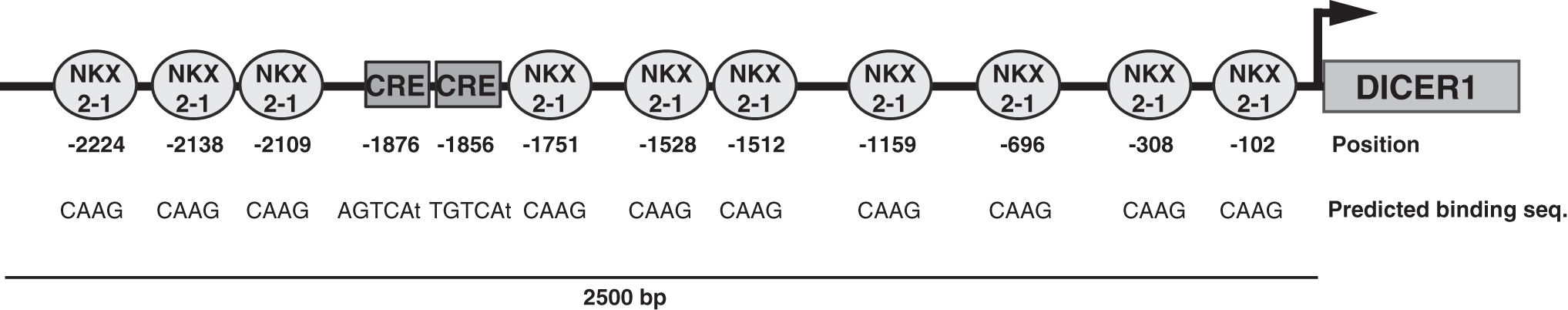
Schematic representation of the promoter region of human DICER1 showing predicted NKX2-1 binding sites and CRE motifs. The relative position from the transcriptional start site (+1) is reported below each transcription factor binding site. Also, the CRE and NKX2-1 consensus sequences are listed below each putative binding site.
TSH regulates DICER1 through the cAMP/CREB-signaling pathway in thyroid cells
The finding of CRE consensus sequences in the DICER1 promoter led us to hypothesize that TSH, through TSHR/cAMP signaling, activates DICER1 to increase DICER1 protein levels. Undifferentiated thyroid tumors lose their response to TSH and its downstream signaling, and this loss in the most undifferentiated tumors could explain, at least in part, the DICER1 downregulation observed in thyroid tumors.
We first studied the regulation of Dicer1 by TSH/cAMP in the TSH-responsive differentiated rat thyroid cell line PCCl3 (22). Cells were starved and maintained in 4H medium for 48 hours and then stimulated with TSH for 2, 14, or 24 hours. We observed that TSH significantly increased Dicer1 mRNA levels at the three time points studied (Fig. 2A). This effect was mimicked by forskolin and inhibited by H89 (Fig. 2B), suggesting that TSH induces Dicer1 mRNA and protein levels via cAMP/PKA signaling. As the CREB transcription factor is a major final effector of cAMP/PKA signaling, we assessed its involvement in TSH-induced DICER1 expression by silencing its expression in PCCl3 cells. As shown in Figure 2C, inhibiting CREB expression significantly reduced TSH induction of DICER1. These results indicate that TSH-dependent stimulation of DICER1 expression is mediated, at least partly, by CREB. Supporting this, an analysis of the mRNA levels of CREB and DICER1 in 496 human tumors from the TCGA database revealed a significant positive correlation between the two (Fig. 2D). Indeed, both CREB and DICER1 were downregulated in PTC samples relative to normal samples, with a fold change of 0.87 and 0.56, respectively (Supplementary Fig. S1A) (3).
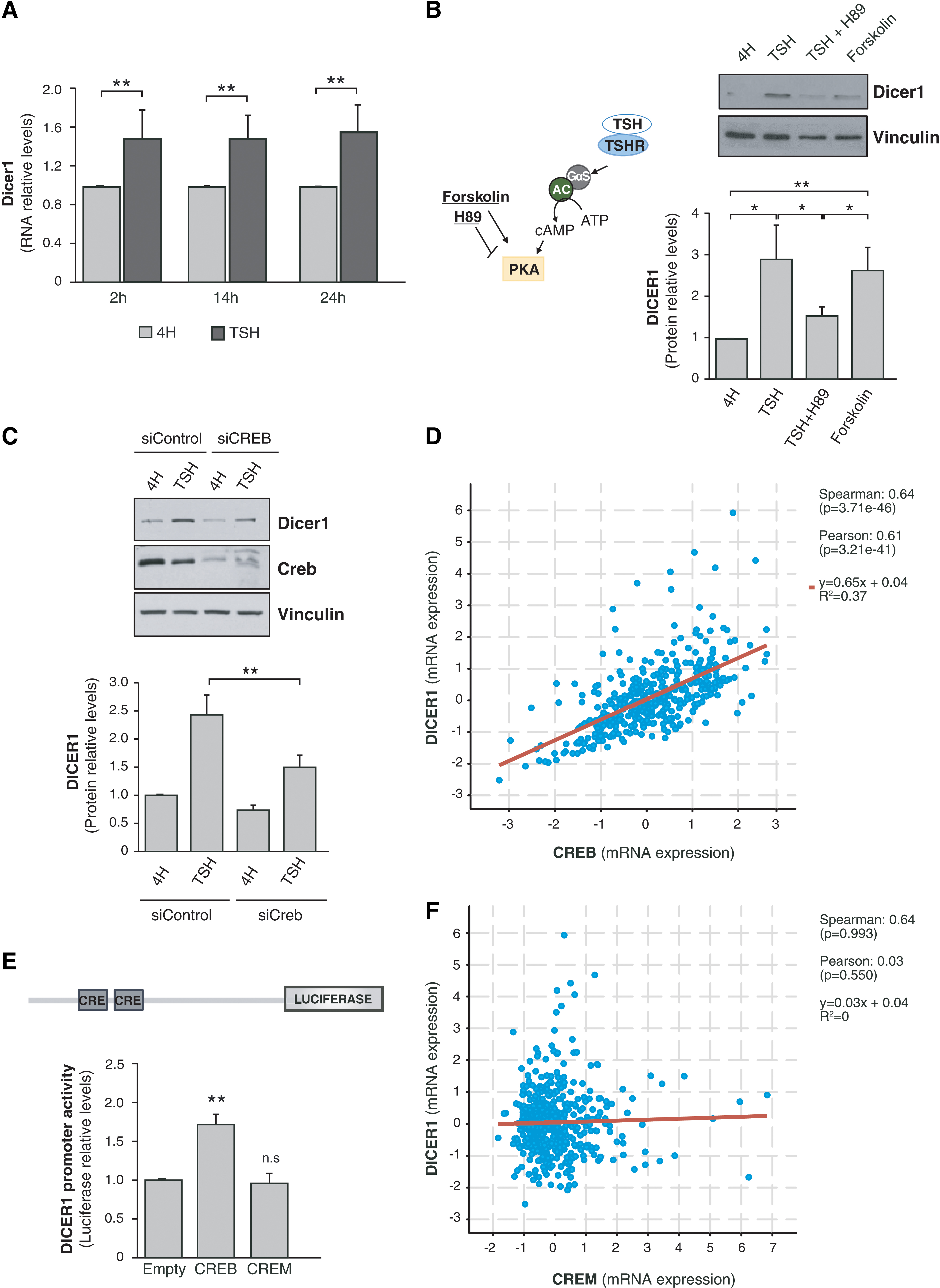
TSH and CREB induce DICER1 promoter activity. (
We next analyzed the transcriptional control of DICER1 by CREB, finding that CREB overexpression activated a human DICER1 promoter construct (Fig. 2E). By contrast, the overexpression of CREM, a transcription factor that also binds CRE elements present in the promoters of cAMP-regulated genes, failed to activate the promoter (Fig. 2E). This latter observation is in good agreement with the lack of correlation between CREM and DICER1 mRNA levels in human thyroid tumors (Fig. 2F).
Existence of a feedback loop between NKX2-1 and DICER1
In silico analysis for thyroid transcription factor binding sites in the human DICER1 proximal promoter predicted several NKX2-1 consensus binding sites (Fig. 1). The transcription factors NKX2-1, PAX8, and FOXE1 are key developmental genes that define the differentiated phenotype of thyroid cells. As cell differentiation is disrupted along thyroid cancer progression and DICER1 is downregulated in thyroid cancer, we next examined the possible transcriptional regulation of DICER1 by NKX2-1 by assessing the functionality of NKX2-1 over the DICER1 promoter. We found that transfected NKX2-1 activated the whole −2500 bp DICER1 promoter in HeLa cells (Fig. 3A). To determine the more critical DICER1 promoter regions for NKX2-1-mediated activation, we made serial deletions of the DICER1 promoter (−1866 − 1250, and −625 bp), finding that the greatest luciferase activity occurred in the pDICER-1250-Luc construct containing the four predicted NKX2-1 binding sites closest to the transcription start site (Fig. 3A), suggesting that the proximal region is responsible for NKX2-1 transcriptional activity. We validated this using ChIP quantitative polymerase chain reaction assays with specific primers to amplify the different NKX2-1 binding sites within the DICER1 promoter. As shown in Figure 3B, NKX2-1 bound to all the predicted sites, with the maximum fold enrichment at sites located at positions −696 and −1159. The specific NKX2-1 binding at position −696 was confirmed by EMSA (Supplementary Fig. S1B).
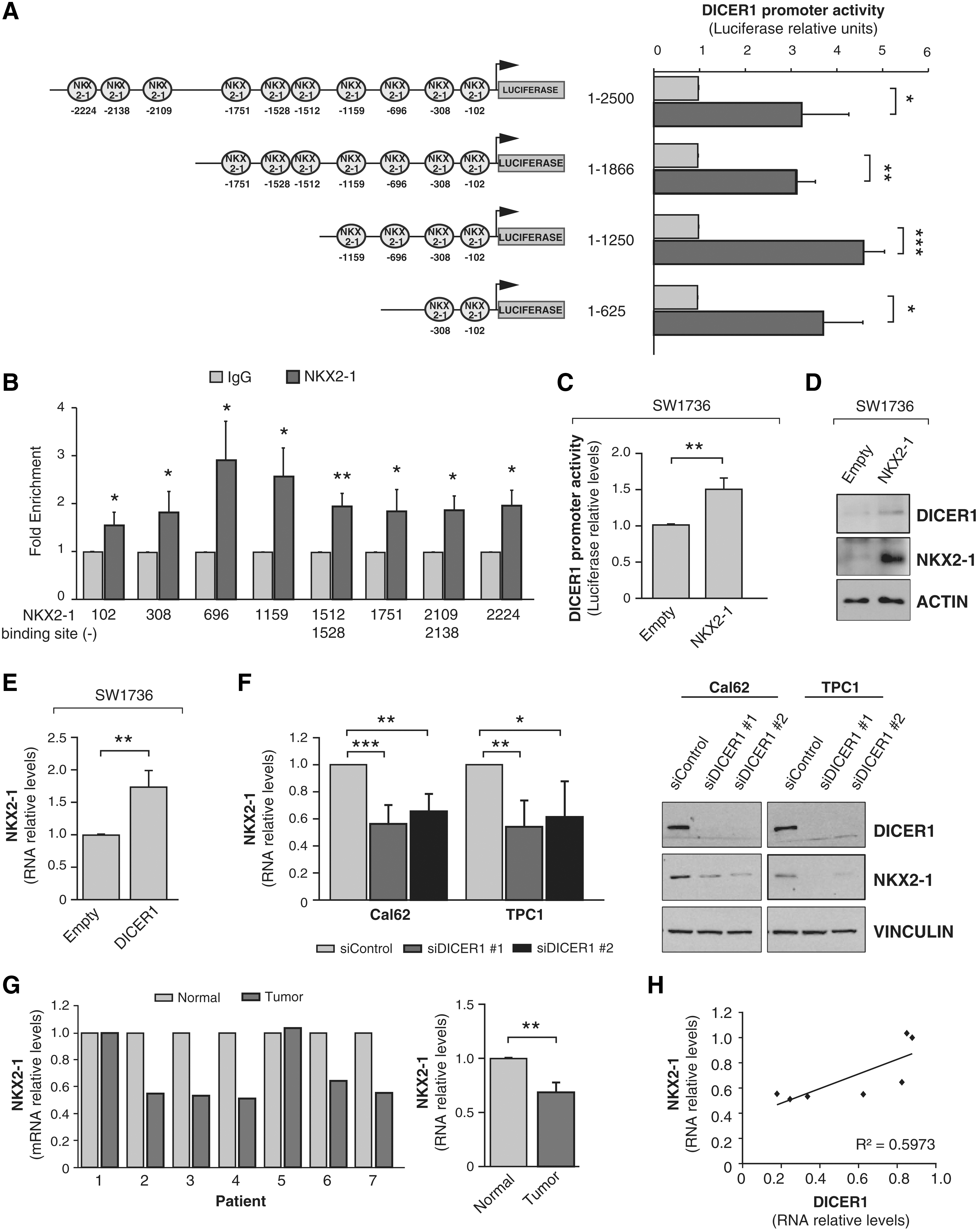
NKX2-1 increases DICER1 promoter activity and DICER1 induces and correlates with NKX2-1 levels. (
NKX2-1 activation of the DICER1 promoter was also reproduced in the ATC cell line SW1736 (Fig. 3C). In addition, overexpression of NKX2-1 increased DICER1 protein levels in SW1736 cells (Fig. 3D), overall confirming the functionality of the NKX2-1 binding sites in the DICER1 promoter, and pointing to a role for NKX2-1 in DICER1 transcription.
We next performed the opposite experiment and overexpressed DICER1 in SW1736 cells, finding a significant increase in NKX2-1 mRNA levels (Fig. 3E). As expected, DICER1 silencing in the ATC lines Cal62 and TPC1 using two different siRNAs significantly decreased NKX2-1 mRNA and protein levels compared with nonsilenced cells (Fig. 3F). These data suggest a role for DICER1 in thyroid differentiation and the existence of a positive feedback loop between DICER1 and NKX2-1.
We confirmed some of these in vitro findings in human thyroid tumors. We observed that NKX2-1 expression was lower in thyroid tumor tissue than in paired normal tissue (Fig. 3G), which is consistent with the described loss of differentiation. We previously described low levels of DICER1 in the same tumors (3). Analysis of DICER1 expression values together with those described here for NKX2-1 revealed a positive correlation (R 2 = 0.5973) between both genes (Fig. 3H).
PAX8 cooperates with NKX2-1 and CREB to regulate DICER1
PAX8 is considered the main transcription factor for thyroid cell differentiation (23). In silico analysis of PAX8 consensus binding sites failed to detect any in the DICER1 proximal promoter, and PAX8 was unable to enhance DICER1 promoter activity in overexpression experiments in HeLa cells (Fig. 4A). Nevertheless, PAX8 cooperated with NKX2-1 and CREB, individually and together, to significantly increase DICER1 promoter activity (Fig. 4A). These data reinforce the idea that in a differentiated state, regulated by TSH, the expression of all three transcription factors increases DICER1 expression, whereas the loss of differentiation in thyroid cancer leads to the impairment of DICER1 transcription, explaining the low levels of this thyroid tumor suppressor.
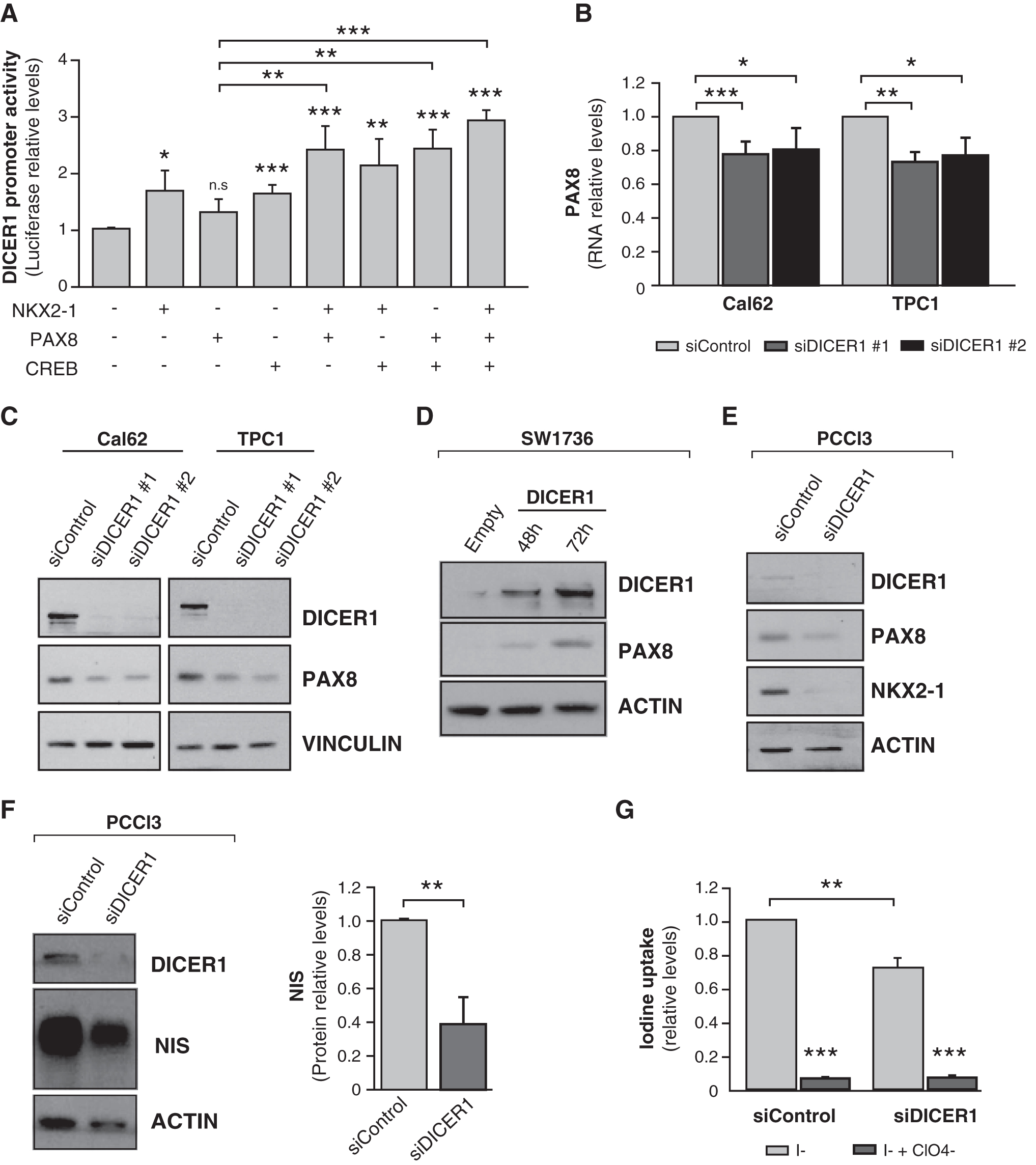
PAX8 cooperates to induce DICER1 transcription, which in turn increases PAX8 and NIS expression. (
DICER1 regulates the levels of the main differentiation transcription factors, leading to loss of NIS expression and iodine uptake
The positive feedback loop between DICER1 and NKX2-1 points to a role for DICER1 in thyroid differentiation. Two previous studies described that conditional deletion of Dicer1 in thyroid follicular cells during mouse development leads to a strong reduction in the expression of Nkx2-1 and Pax8 (17), and in Nis in adult mice (18). We thus examined the link between DICER1 and thyroid differentiation markers. Silencing of DICER1 in Cal62 and TPC1 cell lines decreased PAX8 mRNA (Fig. 4B) and protein (Fig. 4C) expression, whereas DICER1 overexpression had the opposite effect on PAX8 protein expression (Fig. 4D), confirming the involvement of DICER1 in PAX8 regulation.
We studied the expression of NKX2-1, PAX8, and DICER1 in the three thyroid cancer cell lines concurrently. We found a general concordance between the three (Supplementary Fig. S2), with the highest levels in the TPC1 papillary cell line, the most differentiated, supporting a regulatory network between these factors.
A major target of NKX2-1 and PAX8 is the NIS, which is the hallmark of thyroid differentiation. NIS expression and, by extension, function are necessary for the correct synthesis of thyroid hormone and for the diagnostic and therapeutic use of radioiodine in thyroid cancer (24). Using PCCl3 cells, we confirmed that silencing of Dicer1 resulted in a decrease in Nkx2-1 and Pax8 levels (Fig. 4E). NIS expression was significantly decreased in Dicer1-silenced PCCl3 cells (fold change 0.378) (Fig. 4F). This was concomitant with a loss of radioiodine uptake (Fig. 4G), overall suggesting a critical role for DICER1 in maintaining a differentiated thyroid phenotype.
Discussion
Malignancy and differentiation status are inversely correlated. Thyroid cell differentiation is driven by the coordinated action of NKX2-1, PAX8, FOXE1, and HHEX transcription factors, which together regulate the thyroid-specific proteins responsible for thyroid hormone biosynthesis (12,13). Among these, the NIS is essential for treatment of thyroid cancer because radioiodine therapy depends on NIS expression and its correct location in the basolateral membrane of thyroid follicular cells (24,25). Likewise, the active response to TSH via TSHR/cAMP-mediated signaling in differentiated thyroid tumors is critical because TSH is used to induce radioactive iodine uptake (26,27).
Loss of thyroid-specific proteins and differentiation markers is common in thyroid carcinogenesis. Several studies have reported the impaired expression of thyroid transcription factors such as NKX2-1 and PAX8 in thyroid carcinomas, suggesting that their deregulation is pivotal for the initiation and progression of thyroid neoplasms (28 –31). Interestingly, researchers at TCGA developed a thyroid differentiation score based on the expression of several genes, and found an inverse correlation between thyroid transcription factors and tumor malignancy (32).
We recently demonstrated that DICER1 downregulation in thyroid cancer cells correlates with an increase in proliferation, migration, and invasion (3). However, no study had examined the relationship between DICER1 and differentiation in thyroid cancer. The findings that DICER1 is related to thyroid differentiation during thyroid embryonic development (17,18), and that development and cancer share multiple processes, prompted us to analyze the regulation of DICER1 by the main thyroid transcription factors, and the implication of DICER1 downregulation for dedifferentiation in thyroid cancer.
Bioinformatic analysis revealed ten NKX2-1 binding sites and two CREs within the DICER1 promoter and functional analyses demonstrated that TSH via cAMP regulates the expression of DICER1 in differentiated thyroid cells, an effect mediated by CREB. This might indicate that as the response to TSH is lost in tumors, the expression of CREB, and consequently that of DICER1, is also lost. This prediction is supported by the TCGA data, showing a downregulation of DICER1 and CREB expression in tumors and a correlation between them.
We found that NKX2-1 induces DICER1 promoter activity in heterologous assays in HeLa cells and, more importantly, in ATC cells (SW1736). Interestingly, inhibition analysis revealed that DICER1 is positively associated with NKX2-1 levels. The existence of a positive feedback loop between NKX2-1 and DICER1 might have important implications in thyroid cancer, as is supported by the positive correlation between both proteins in a series of patient samples analyzed here.
Akin to NKX2-1, PAX8 plays a key role in thyroid differentiation. However, neither the PAX8 consensus nor the degenerated sequence (33) was evident in the proximal DICER1 promoter. While PAX8 by itself failed to activate DICER1 transcription, it cooperated additively with both NKX2-1 and CREB, which is consistent with the observation of an interaction between NKX2-1 and PAX8 that synergistically activates transcription (34). In addition, CRE binding proteins interact with PAX8 to regulate NIS transcription (35,36). These data strongly suggest that the coordinated action of the three transcription factors, under the control of TSH, triggers DICER1 expression in differentiated thyroid cells. In turn, loss of these differentiation factors reduces DICER1 expression and results not only in dedifferentiation but also enhanced cell aggressiveness (3).
Prior studies showed that the absence of Dicer1 does not significantly affect the expression of Nkx2-1 and Pax8 at the end of embryonic development and at the beginning of birth (18); however, there is a decrease in both transcription factors in adults (17). These mice show severe hypothyroidism and loss of Tshr, TPO, and Nis expression with signs of neoplastic alterations.
Interestingly, among the cell lines studied, we observed that TPC1 cells have the highest levels of DICER1, PAX8, and NKX2-1. This fits with our hypothesis, as TPC1 is a papillary cell line and is therefore more differentiated than the anaplastic cell lines (Cal62 and SW1736). Also worthy of note is the finding that SW1736 cells show lower levels of DICER1, PAX8, and NKX2-1 than Cal62, suggesting that it is even less differentiated. This might be because SW1736 harbors mutations in BRAFV600E, whereas Cal62 harbors a mutation in KRASG12R. The BRAF mutation is believed to confer higher aggressiveness and is associated with a lower differentiation score (32). Importantly, there is a concordance between the three factors in the three cell lines; the highest levels of PAX8 and NKX2-1 are found in cells with the highest levels of DICER1. These results support our hypothesis and reveal a regulatory network between the three factors.
We found that PAX8 and NKX2-1 levels are significantly reduced in DICER1-silenced human thyroid tumor cells, whereas the opposite is seen in DICER1 overexpressing cells. Similar results were obtained in Dicer1-silenced differentiated PCCl3 cells. Interestingly, the levels of Nis, the main target of Pax8, also decreased after Dicer1 silencing, resulting in a significant reduction in iodine uptake. We previously reported low DICER1 mRNA levels in thyroid tumors (3). This agrees with data from the Human Protein Atlas, in which DICER1 protein levels are decreased in thyroid tumor samples. Specifically, all control samples (from normal thyroid gland) show positive (although weak) staining in the majority (75%) of cells, whereas staining is weak or absent in thyroid carcinoma tissue and the fraction of stained cells is less than 25% in three of four samples. We speculate that the low DICER1 levels lead to differentiated tumors with a markedly decreased expression of NIS, making radioiodine treatment futile. Thus, the recovery of thyroid transcription factors should ultimately be effective for DICER1 expression and for functional NIS activity.
In summary, we demonstrate that the miRNA biogenesis enzyme DICER1, acting as a tumor suppressor, is finely regulated in thyroid cells. Our results support the following scenario (Fig. 5): TSH, the main hormone controlling proliferation and differentiation in thyroid follicular cells, stimulates DICER1 expression via TSHR/cAMP/PKA, CREB, NKX2-1, and PAX8 to cooperatively regulate DICER1, and a positive feedback loop between NKX2-1 and DICER1 implicates a role for DICER1 in thyroid differentiation. Indeed, DICER1 upregulates NKX2-1 and PAX8 levels inducing NIS expression and function, thereby increasing iodine uptake. Most of our findings were supported by an exhaustive analysis of TCGA and human tumoral samples, allowing us to conclude that DICER1 downregulation in thyroid tumors is mediated, at least partly, through the impairment of its transcription.
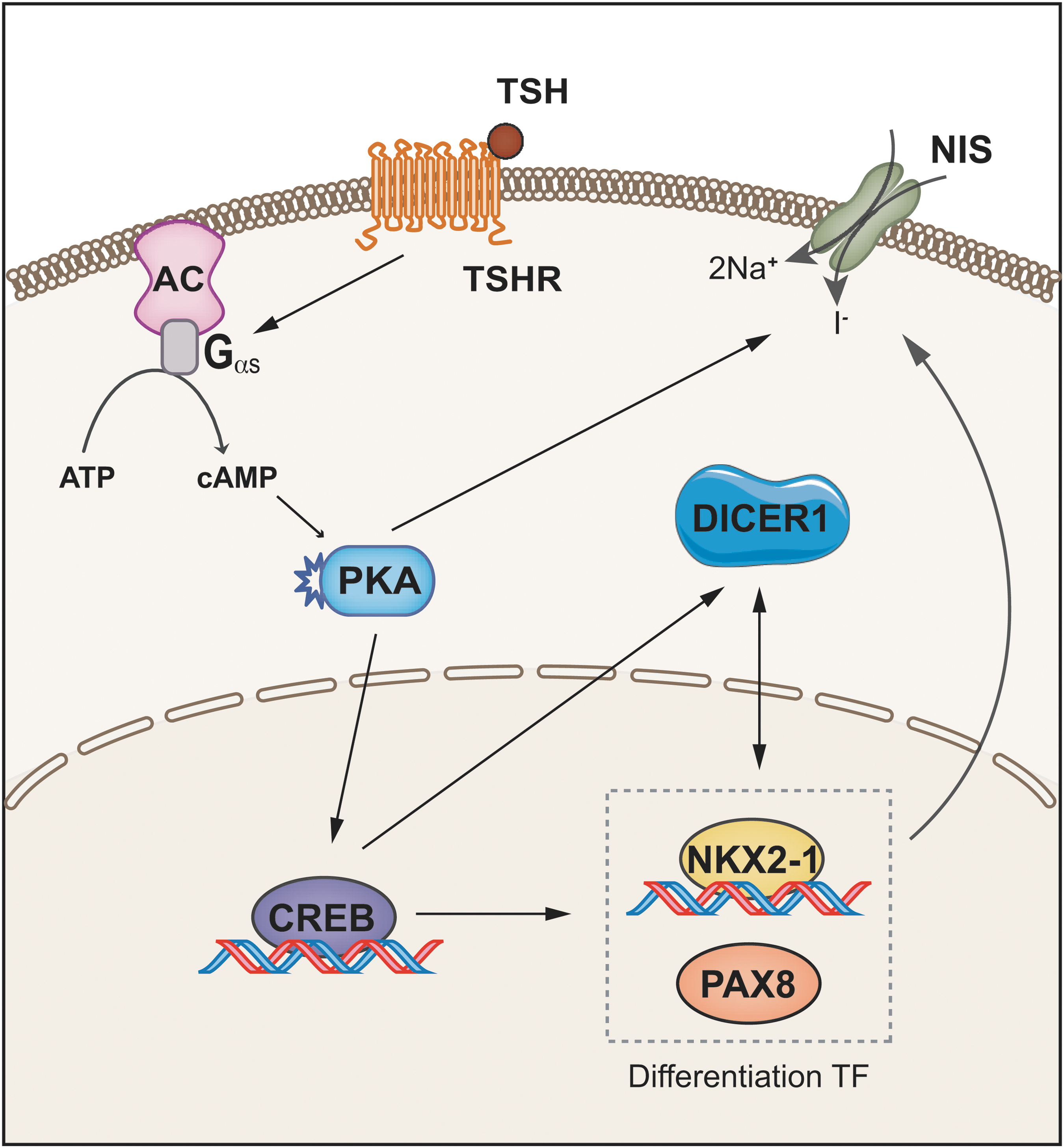
Schematic summary. TSH acting through its G protein-coupled receptor induces the cAMP/PKA/CREB pathway, which regulates the transcription of DICER1. A feedback loop exists between DICER1 and the thyroid transcription factors NKX2-1 and PAX8. Both transcription factors, in cooperation, induce the transcription of DICER1, which in turn controls PAX8 and NKX2-1 levels, inducing NIS expression and iodine uptake. TSHR, TSH receptor. Color images are available online.
Footnotes
Acknowledgments
We thank Dr. Kenneth McCreath for his comments on the article and Dr. Garcilaso Riesco-Eizaguirre, Hospilal Universitario de Mostoles, Madrid) for providing thyroid tumor samples and clinical information of the patients.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants SAF2016-75531-R, Fondo Europeo de Desarrollo Regional, and PID2019-105303RB-I00/AEI/10.13039/501100011033 from Ministerio de Ciencia e Innovación (MICIN), Spain, B2017/BMD-3724 from Comunidad de Madrid, and GCB14142311CRES from Fundación Española Contra el Cáncer (AECC). J.R.-M. holds an FPU fellowship from MECD (Spain).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1