
Abstract
Background:
Several single-nucleotide polymorphisms (SNPs) are known to increase the risk of Hashimoto's thyroiditis (HT); such SNPs reside in thyroid-specific genes or in genes related to autoimmunity, inflammation, and/or cellular defense to stress. The transcription factor Nrf2, encoded by NFE2L2, is a master regulator of the cellular antioxidant response. This study aimed to evaluate the impact of genetic variation in NFE2L2 on the risk of developing HT.
Methods:
In a case–control candidate gene association study, functional SNPs in the NFE2L2 promoter (rs35652124, rs6706649, and rs6721961) were examined either as independent risk factors or in combination with a previously characterized HT risk allele (rs28665122) in the gene SELENOS, encoding selenoprotein S (SelS). A total of 997 individuals from the north of Portugal (Porto) were enrolled, comprising 481 HT patients and 516 unrelated healthy controls. SELENOS and NFE2L2 SNPs were genotyped using TaqMan® assays and Sanger sequencing, respectively. Odds ratios (ORs) were calculated using logistic regression, with adjustment for sex and age. Expression of SelS was analyzed by immunohistochemistry in thyroid tissue from HT patients and control subjects. Molecular interactions between the Nrf2 and SelS pathways were investigated in thyroid tissues from mice and in rat PCCL3 thyroid follicular cells.
Results:
When all three NFE2L2 SNPs were considered together, the presence of one or more minor alleles was associated with a near-significant increased risk (OR = 1.43, p = 0.072). Among subjects harboring only major NFE2L2 alleles, there was no increased HT risk associated with heterozygosity or homozygosity for the SELENOS minor allele. Conversely, in subjects heterozygous or homozygous for the SELENOS risk allele, the presence of an NFE2L2 minor allele significantly increased HT risk by 2.8-fold (p = 0.003). Immunohistochemistry showed reduced expression of SelS in thyroid follicular cells of HT patients. In Nrf2 knockout mice, there was reduced expression of SelS in thyroid follicular cells; conversely, in PCCL3 cells, reducing SelS expression caused reduced activity of Nrf2 signaling.
Conclusions:
The NFE2L2 promoter genotype interacts with the SELENOS promoter genotype to modulate the risk of HT in a Portuguese population. This interaction may be due to a bidirectional positive feedback between the Nrf2 and SelS pathways.
Introduction
Hashimoto's thyroiditis (HT) is a chronic autoimmune disease with an annual incidence of 0.3–1.5/1000 and a prevalence of up to 10% in the general population (1,2). It is characterized by thyroid inflammation due to expansion of thyroid antigen-specific lymphocytes that infiltrate the gland and production of thyroid autoantibodies, leading to gradual destruction of the thyroid parenchyma and often subsequent hypothyroidism (3). HT is a multifactorial disorder whose pathogenesis involves genetic and environmental predisposing factors (1,3 –5). Twin studies estimate its heritability at 70% (4), and several single-nucleotide polymorphisms (SNPs) are known to increase the risk of HT. Such SNPs reside either in thyroid-specific genes (e.g., TSHR and TG) (6,7) or in genes related to autoimmunity, inflammation, and/or cellular mechanisms of defense to stress (e.g., IL-1β, IL-6, and TNF) (8,9).
We have previously identified one such HT susceptibility gene, SELENOS, which encodes a member of the selenoprotein family, selenoprotein S (SelS) (10). SELENOS is expressed in multiple tissues, including thyroid follicular cells, and the protein it encodes is involved in cellular stress responses as a mediator of endoplasmic reticulum (ER)-associated protein degradation (ERAD) of misfolded proteins (11), as well as in immune and inflammatory processes (12). We previously showed that a functional SNP in the SELENOS promoter (rs28665122, −105G/A) is associated with increased risk of HT (10). Specifically, heterozygosity or homozygosity for the minor allele (GA or AA, respectively) was significantly more frequent in HT patients than in healthy controls, increasing the risk of HT by more than twofold (10). In luciferase reporter gene assays, the allele A shows reduced promoter transcriptional activity (12), which suggests that reduced SELENOS transcription may influence the proinflammatory cytokine profiles observed in thyroid autoimmunity and/or the proteostatic responses of thyroid follicular cells, thereby predisposing to HT.
We hypothesized that additional risk factors for HT might reside in cellular systems that regulate or cross talk to selenoproteins and mediate complementary and/or overlapping cellular stress responses. One such candidate is NFE2L2, which encodes the nuclear factor-erythroid 2-related transcription factor 2 (NFE2L2 or Nrf2), a member of the cap'n'collar leucine zipper family (13). Nrf2 is known to regulate the basal and inducible expression of a battery of cell protective genes that encode antioxidant and detoxification enzymes, proteasome subunits, and other proteostasis mediators (14). In the absence of oxidative stress, Nrf2 binds to its cytoplasmic inhibitor Kelch-like ECH-associated protein 1 (Keap1), which targets Nrf2 for polyubiquitination and proteasomal degradation (13,15). Keap1 also functions as a sensor of oxidants and electrophiles, which react with its redox-sensitive cysteines (16,17). Oxidative stressors abolish the inhibition of Nrf2 by Keap1; Nrf2 then accumulates in the nucleus where it transcriptionally activates protective genes through antioxidant response elements (AREs) in their regulatory sequences (13). Studies in different tissues have shown that Nrf2 regulates hundreds of genes, including several selenoproteins (13,14). In addition, multiple lines of evidence indicate that the selenoprotein system and the Nrf2 system can compensate for each other in antioxidant stress responses, such that the simultaneous disruption of both systems is particularly deleterious to proteostasis (18 –23).
The human NFE2L2 gene maps to chromosome 2q31 and consists of 5 exons and 4 introns. Three functional SNPs have been identified in the NFE2L2 promoter (24): rs35652124, −214A>G; rs6706649, −212G>A; and rs6721961, −178A>C—nomenclature according to a study (25). Each of these SNPs has been found to affect the expression level of Nrf2, with the minor alleles associated with a lower promoter activity (26,27). These polymorphisms have been associated with several diseases related to oxidative stress (25), such as acute lung injury (26), asthma (28), Parkinson's disease (29), Alzheimer's disease (30), and others, including autoimmune diseases such as systemic lupus erythematosus (31) and vitiligo (32).
Autoimmune thyroid disease has also been associated with systemic oxidative stress (33,34); however, the potential role of NFE2L2 SNPs in susceptibility to autoimmune thyroid disease has not been addressed. Interestingly, Nrf2 knockout (KO) mice are known to develop an age-related multiorgan autoimmune syndrome (35 –37), but it has not been reported whether the thyroid gland is also affected. Recently, we showed that Nrf2 positively controls thyroglobulin expression while limiting its iodination in the thyroid of mice (38). We also showed that Nrf2 positively controls the thyroidal expression of antioxidant genes, including those encoding the selenoproteins glutathione peroxidase 2 (Gpx2) and thioredoxin reductase 1 (Txnrd1) (38), which are both known to have important roles in follicular cell homeostasis (39,40). However, the involvement of Nrf2 in autoimmune thyroid disease has not been investigated. For these reasons, we investigated whether the known functional NFE2L2 promoter SNPs, either individually or in combination with each other and/or with the known SELENOS promoter functional SNP, modulate the risk of HT.
Materials and Methods
Study subjects
Associations between the three NFE2L2 promoter SNPs and the SELENOS promoter SNP and the risk of HT were assessed in a case–control study using cohorts that have been previously described in detail (10). Briefly, a total of 997 subjects were enrolled; these included 481 genetically unrelated HT patients (441 females and 40 males; mean age 55.3 years; range 30–62 years) enrolled at the Department of Endocrinology of Hospital of S. João (Porto, Portugal). The control group comprised 516 subjects (479 females and 37 males; mean age 46.3 years; range 18–83 years) recruited among unrelated healthy blood donors. Patients were enrolled between 2007 and 2013; diagnosis was made based on the presence of positive serum autoantibodies against thyroid peroxidase and/or thyroglobulin and negative serum autoantibodies against the thyrotropin (TSH) receptor, according to methods applied before or after March 2009 in the Department of Clinical Pathology of Hospital of S. João, in conjunction with a typical ultrasound pattern of the thyroid parenchyma (i.e., diffuse hypoechogenicity and heterogeneity). Subjects with a reported history of thyroid cancer and/or prior thyroid surgery were excluded. The biochemical characteristics of the patient cohort have been reported previously (10); all patients were treated with levothyroxine for HT-related hypothyroidism. The control group, healthy blood donors without apparent infectious and/or chronic disorders, consisted of permanent residents in the area serviced by the Hospital of S. João, selected during the assembly of the EpiPorto cohort (41). The study was authorized by the Hospital of S. João Ethics Committee and informed consent was obtained from all participants.
Formalin-fixed paraffin-embedded thyroid tissue samples from control subjects (n = 10) or patients with HT (n = 8), collected during thyroidectomy performed for other indications (notably benign goiter), were obtained from the archives of Lausanne University Hospital, with authorization from the Ethics Committee of the Canton of Vaud (study no. 2017-00944).
Genotyping
Genomic DNA from patients and controls was isolated from blood using standard proteinase K digestion with phenol/chloroform extraction. For the NFE2L2 SNPs rs35652124, rs56706649, and rs6721961, polymerase chain reaction (PCR) amplifications were performed using forward primer 5′-CTGCGCTTTGGTGGGAAG-3′ and reverse primer 5′-TGGAGTTGCAGAACCTTGC-3′. Briefly, PCR amplifications were performed in 25 μL volume reactions containing 100 ng genomic DNA, 100 μM of each dNTP, 0.1 μg of each primer, 1 × GoTaq® Flexi Buffer (Promega, Madison, WI), 2.5 mM MgCl2, and 0.15 U of GoTaq G2 Flexi DNA Polymerase (Promega). Reaction mixtures were submitted to 35 amplification cycles preceded by a Taq activation step at 95 °C for 5 minutes; each cycle comprised a denaturation step at 95 °C for 30 seconds, annealing at 59 °C for 30 seconds, and extension at 72 °C for 45 seconds followed by a final extension at 72 °C for 5 minutes. PCR products were then purified by enzymatic digestion with exonuclease I (Thermo Fisher Scientific, Waltham, MA) and shrimp alkaline phosphatase (Thermo Fisher Scientific) and sequenced using the BigDye® Terminator v3.1 Kit (Applied Biosystems, Foster City, CA) and an ABI Prism 3100 Genetic Analyzer (Perkin-Elmer, Waltham, MA). In a randomly selected 10% of the samples, PCR and sequencing were performed twice to verify the reproducibility of the technique. The number of subjects successfully genotyped for each NFE2L2 SNP differed slightly according to the success of the respective sequencing reaction assay. Genotyping of SELENOS rs28665122 in the same subjects had been performed previously using TaqMan® assays (10).
Mice
C57BL/6J Nrf2+/− mice, originally developed by Prof. M. Yamamoto (42), were obtained from RIKEN BRC (Tsukuba, Japan). Mice were housed in the animal facility of the University of Patras Medical School in temperature-, light-, and humidity-controlled rooms with a 12-hour light/dark cycle. All animal procedures were approved by the local institutional review board and were in accordance with the European Commission Directive 86/609/EEC. Generation of wild-type (WT) and KO (Nrf2-KO) mice and tissue collection has been previously described (38).
Cell culture
PCCL3 cells, a clonal rat thyroid cell line (43), were cultured in Coon's modified Ham's F-12 under conditions described previously (38). Culture media and supplements were all from Sigma-Aldrich (St-Louis, MO). Generation of PCCL3 Nrf2-KO and PCCL3 Keap1-KO cells using CRISPR/Cas9 technology has been previously described (38).
Cell viability assay
The CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega) was used according to the manufacturer's protocol to determine the number of viable cells in culture based on the quantification of adenosine triphosphate. Briefly, cells were lysed by adding 100 μL CellTiter-Glo reagent to each well and the luminescence intensity was measured 10 minutes later on a NOVOstar multimode reader (BMG Labtech, Ortenberg, Germany). Results were expressed as fold change over the untreated control sample.
RNA isolation and real-time PCR
Total RNA isolation and real-time PCR (RT-PCR) from mouse tissues and from cell lines were performed as previously described (38). Melt curve analysis was used to confirm gene-specific amplification. Relative gene expression was calculated by the comparative cycle threshold method using PPIA as reference gene for thyroid and liver tissues and RPL19 for PCCL3 cells. Primers for RT-PCRs are shown in Supplementary Tables S1,S2,S3.
Protein isolation, Western immunoblotting, and immunohistochemistry
Total protein isolation and Western blots were performed as previously described (38). The following primary antibodies were used: anti-SelS (HPA010025, 1:3000; Sigma-Aldrich), produced in rabbit, a Prestige Antibody, developed and validated by the Human Protein Atlas (HPA) project; anti-vinculin (SAB4200080, 1:5000; Sigma-Aldrich); and anti-β-actin (#4967, 1:5000; Cell Signaling, Danvers, MA). Secondary antibodies were anti-mouse HRP (#7076) and anti-rabbit HRP (#7074) both from Cell Signaling (1:5000).
Immunohistochemistry was performed on slices from formalin-fixed paraffin-embedded human or mouse thyroid tissues as previously described (38,44) using the aforementioned primary antibody against SelS. Specific binding was detected with the Envision Kit (Dako), and the color reaction was visualized with 3,3′-diaminobenzidine. For negative controls, blocking solution was added instead of a primary antibody.
RNA interference
Silencing of SelS was performed using the DsiRNA TriFECTa Kit (cat. no. 1
Cell treatments
Unless otherwise stated, all chemicals were from Sigma-Aldrich. The following compounds were used for cell treatments: sulforaphane (SLF, R-1-isothiocyanato-4-methylsulfinylbutane) was used as Nrf2 activator (45). Oxidative stress conditions were induced using menadione (2-methyl-1,4-naphthoquinone, vitamin K3), a redox-cycling agent widely used in the literature to induce oxidative damage (46). In preliminary experiments, various ranges of concentrations and treatment times were tested to identify conditions where the viability of WT cells remained unaffected. Finally, an 8-hour treatment at a concentration of 20 nM was used in all experiments. To induce ER stress, cells were treated with the protein glycosylation inhibitor tunicamycin A (TN) at a concentration of 0.5 mg/mL (47).
Promoter analysis, cell transfections, and reporter assays
The promoter sequence of the human SELENOS gene was downloaded from the EPDnew database (promoter ID: VIMP_1,
Transient transfections of cells with the different promoter reporter constructs were performed in 48-well plates cultured in complete medium using a 1:2 ratio of DNA and jetPRIME transfection reagent, respectively. In all transfections, the pEGFP-N1 plasmid (Clontech, Mountain View, CA) was included for monitoring transfection efficiency and normalizing luciferase activities, as previously described (49). Treatment of cells with SLF (5 μM) or TN (0.5 μg/mL) or vehicle (DMSO or dimethylsulfoxide) was performed 24 hours after transfection. Cells were lysed 48 hours after transfection using the passive lysis buffer (Promega), and green fluorescent protein (GFP) fluorescence was measured using a NOVOstar multimode reader with an excitation wavelength of 480 nm and an emission wavelength of 520 nm. The GFP fluorescence background of untransfected cells was used for blank measurements. After measuring GFP fluorescence, luciferase activities were measured in the same plates using the luciferase assay system (Promega) according to the manufacturer's protocol. Luciferase activities were normalized to the respective GFP fluorescence measurements, and relative luciferase activities were expressed as fold change over control.
Statistics
For the genetic studies, statistical analyses were performed using SPSS v.25.0 (IBM, Armonk, NY). The Hardy–Weinberg equilibrium was evaluated using a chi-square test. Comparison of genotype frequencies between HT patients and controls was performed using unconditional logistic regression. Odds ratios (ORs) with the respective 95% confidence intervals (CIs) for association of the SELENOS and NFE2L2 polymorphisms with HT were calculated for the genotypic and dominant models of inheritance with adjustment for sex and age. Controlling for the effects of multiple testing was performed by the false discovery rate (FDR) method (50).
For ordinal data (staining intensity in control vs. Hashimoto's samples), the Kruskal–Wallis test was used in GraphPad Prism 8 (GraphPad Software, La Jolla, CA), which was also used to prepare all graphs. For data calculated as normalized ratios [messenger RNA (mRNA), protein, and luciferase quantifications], the BootstRatio application was used; this tool is based on bootstrapping and resampling methods without any assumption on the underlying probability distribution for the data analyzed (51). Whenever the same data were used to test multiple hypotheses, a Bonferroni correction was applied as appropriate.
Results
Distribution of NFE2L2 promoter SNP genotypes and haplotypes and HT risk
In the control group, the genotype frequencies for all three NFE2L2 SNPs (as well as for SELENOS rs28665122) did not deviate significantly from those expected under Hardy–Weinberg equilibrium (all p > 0.05). The impact of each NFE2L2 SNP was first examined individually and independently of the SELENOS SNP. These analyses included subjects successfully genotyped for each NFE2L2 SNP: rs35652124 (403 patients and 514 controls); rs6706649 (403 patients and 543 controls); and rs6721961 (403 patients and 555 controls). On an individual basis, none of the NFE2L2 SNP genotypes showed a significant association with the risk of HT (all p > 0.05; Table 1). A per allele analysis confirmed the absence of statistically significant associations for each of the SNPs: For rs35652124, allele A (reference allele) was present in 75.88% of controls and 73.20% of patients, and allele G was present in 24.12% of controls and 26.80% of patients (OR = 1.15 [CI 0.93–1.42], p = 0.191). For rs6706649, allele G (reference allele) was present in 87.57% of controls and 89.97% of patients, and allele A was present in 12.43% of controls and 13.03% of patients (OR = 1.06 [CI 0.80–1.39], p = 0.700). Finally, for rs6721961, allele C (reference allele) was present in 90.54% of controls and 88.71% of patients, and allele A was present in 9.46% of controls and 11.29% of patients (OR = 1.22 [CI 0.91–1.64], p = 0.192). Finally, when NFE2L2 haplotypes were constructed for patients and controls, four haplotypes with frequencies >1% were identified (Table 2). There was no difference in the frequency of each of these haplotypes between patients and controls (all p > 0.05; Table 2). However, each of these haplotypes was marked by only one NFE2L2 promoter SNP minor allele (Table 2); hence, these results are not more informative than the aforementioned individual analyses (Table 1). In summary, these results indicate that neither the individual NFE2L2 promoter SNPs nor the main respective NFE2L2 promoter haplotypes are independent risk factors for HT.
Frequencies of Individual NFE2L2 Promoter Single-Nucleotide Polymorphism Genotypes in Hashimoto's Thyroiditis (HT) Patients and Controls, and Calculated Risk of HT for Each Genotype
Model adjusted for sex and age.
Versus respective reference group.
Reference groups.
CI, confidence interval; N/A, not applicable; OR, odds ratio; SNP, single-nucleotide polymorphism.
NFE2L2 Promoter Haplotypes and Respective Calculated Risk of Hashimoto's Thyroiditis
Minor alleles are underlined. Model adjusted for sex and age.
By order of position on chromosome 2: rs35652124, rs6706649, and rs6721961.
Reference haplotype.
Combined NFE2L2 promoter genotypes and HT risk
The three functional NFE2L2 SNPs lie close together within 36 bp of the promoter, and reporter gene assays have shown that promoter activity decreases with a higher number of minor alleles (27). Therefore, we next considered the joint impact of all three NFE2L2 SNPs on HT risk. Heterozygosity or homozygosity for any 1, 2, or 3 of the NFE2L2 SNP minor alleles was not significantly associated with increased HT risk (p = 0.072; Table 3). Nevertheless, the fact that the p-value was close to significance prompted us to investigate the possible interaction of the NFE2L2 SNPs with the SELENOS functional SNP known to impact the risk of HT in the same population (10).
Number of Combined NFE2L2 Promoter Single-Nucleotide Polymorphism Minor Alleles and Respective Calculated Risk of Hashimoto's Thyroiditis
Model adjusted for sex and age.
Minor allele heterozygous or homozygous for any NFE2L2 promoter SNPs.
Interactions between individual NFE2L2 and SELENOS promoter SNP genotypes and HT risk
Next, for each of the NFE2L2 SNPs, its effect was assessed in combination with the previously documented SELENOS SNP risk allele (10). These analyses included 586 subjects successfully genotyped for all three NFE2L2 SNPs and for the SELENOS SNP (322 patients and 264 controls). First, each NFE2L2 SNP was considered separately. In subjects not carrying the known SELENOS risk allele (GG homozygotes), heterozygosity or homozygosity for an NFE2L2 SNP minor allele did not modify the risk of HT (all p > 0.05; Tables 4 –6, group 2 vs. group 1). Similarly, in subjects heterozygous or homozygous for the SELENOS risk allele (GA or AA, respectively), heterozygosity or homozygosity for an NFE2L2 SNP minor allele did not modify the risk of HT (all p > 0.05; Tables 4 –6, group 4 vs. group 3). Finally, homozygosity for an NFE2L2 SNP major allele did not abolish the increased HT risk associated with heterozygosity or homozygosity for the SELENOS risk allele (all p > 0.05; Tables 4 –6, group 3 vs. group 1). In summary, these results indicate that when each of the three functional NFE2L2 promoter SNPs is considered individually, none of them either increases or abolishes the risk of HT associated with the SELENOS promoter functional SNP.
Frequencies of the SELENOS rs28665122 and NFE2L2 rs35652124 Single-Nucleotide Polymorphism Combined Genotypes in HT Patients and Controls, and Calculated Risk of HT for Each Genotype
Bold indicates statistical significance.
Model adjusted for sex and age.
Frequencies of the SELENOS rs28665122 and NFE2L2 rs6706649 Single-Nucleotide Polymorphism Combined Genotypes in HT Patients and Controls, and Calculated Risk of HT for Each Genotype
Bold indicates statistical significance.
Model adjusted for sex and age.
Frequencies of the SELENOS rs28665122 and NFE2L2 rs6721961 Single-Nucleotide Polymorphism Combined Genotypes in HT Patients and Controls, and Calculated Risk of HT for Each Genotype
Bold indicates statistical significance.
Model adjusted for sex and age.
Interactions between combined NFE2L2 and SELENOS promoter SNP genotypes and HT risk
Finally, we considered the combined impact of all three NFE2L2 SNPs on the risk of HT associated with the SELENOS promoter functional SNP genotype. In subjects not carrying the known SELENOS risk allele, heterozygosity or homozygosity for any one of the NFE2L2 SNP minor alleles did not modify the risk of HT (p = 0.979; Table 7, group 2 vs. group 1). Moreover, the presence of additional NFE2L2 SNP minor alleles (from 2 to 6 total) also did not modify the risk of HT compared with the presence of a single minor allele (p = 0.879; Table 7, group 3 vs. group 2). These results are consistent with the notion that the NFE2L2 promoter genotype is not an independent risk factor for HT (Tables 1 –3). However, among subjects harboring only major NFE2L2 alleles, heterozygosity or homozygosity for the SELENOS SNP minor allele was not associated with increased risk of HT (p = 0.704; Table 7, group 3 vs. group 1), suggesting a protective effect of the NFE2L2 major alleles. In further support of an interaction effect, in subjects heterozygous or homozygous for the SELENOS risk allele, heterozygosity or homozygosity for any one of the NFE2L2 SNP minor alleles significantly increased the risk of HT by 2.8-fold (p = 0.003; Table 7, group 5 vs. group 4), suggesting a predisposing effect of the NFE2L2 minor alleles. Of note, statistical significance was retained after controlling for the effects of multiple testing using the FDR method. Subjects with additional NFE2L2 SNP minor alleles (from 2 to 6 total) had the same level of risk of HT as those with a single minor allele (p = 0.428; Table 7, group 6 vs. group 5). In summary, these results indicate that when the three NFE2L2 promoter functional SNPs are considered together and in combination with the SELENOS promoter functional SNP, NFE2L2 major alleles abolish the HT risk associated with the SELENOS minor allele, whereas NFE2L2 minor alleles increase the risk.
Interaction Between Number of Combined NFE2L2 Promoter Single-Nucleotide Polymorphism Minor Alleles and SELENOS Minor Alleles, and Respective Calculated Risk of HT
Bold indicates statistical significance.
Model adjusted for sex and age.
Minor allele heterozygous or homozygous for any NFE2L2 promoter SNPs.
Reduced expression of SelS in thyroid follicular cells of Hashimoto's patients
To better understand how the genetic interaction between SELENOS and NFE2L2 may impact the susceptibility to Hashimoto's disease, we first analyzed the expression of SelS protein by immunohistochemistry in thyroid samples from control subjects (n = 10) or patients with HT (n = 8), collected during thyroidectomy performed for other indications (notably benign goiter). Representative immunohistochemical stainings and respective staining intensity scores are shown in Figure 1A, and the distribution of staining intensities among samples is depicted in the histograms shown in Figure 1B. There was a statistically significant lower expression of SelS (p ≤ 0.001) in the thyroid follicular cells of HT compared with those in control samples. Interestingly, despite the reduced expression of SelS in thyroid follicular cells in Hashimoto's samples, a subset of lymphocytes in germinal centers and other lymphocytic infiltrates stained strongly for SelS.
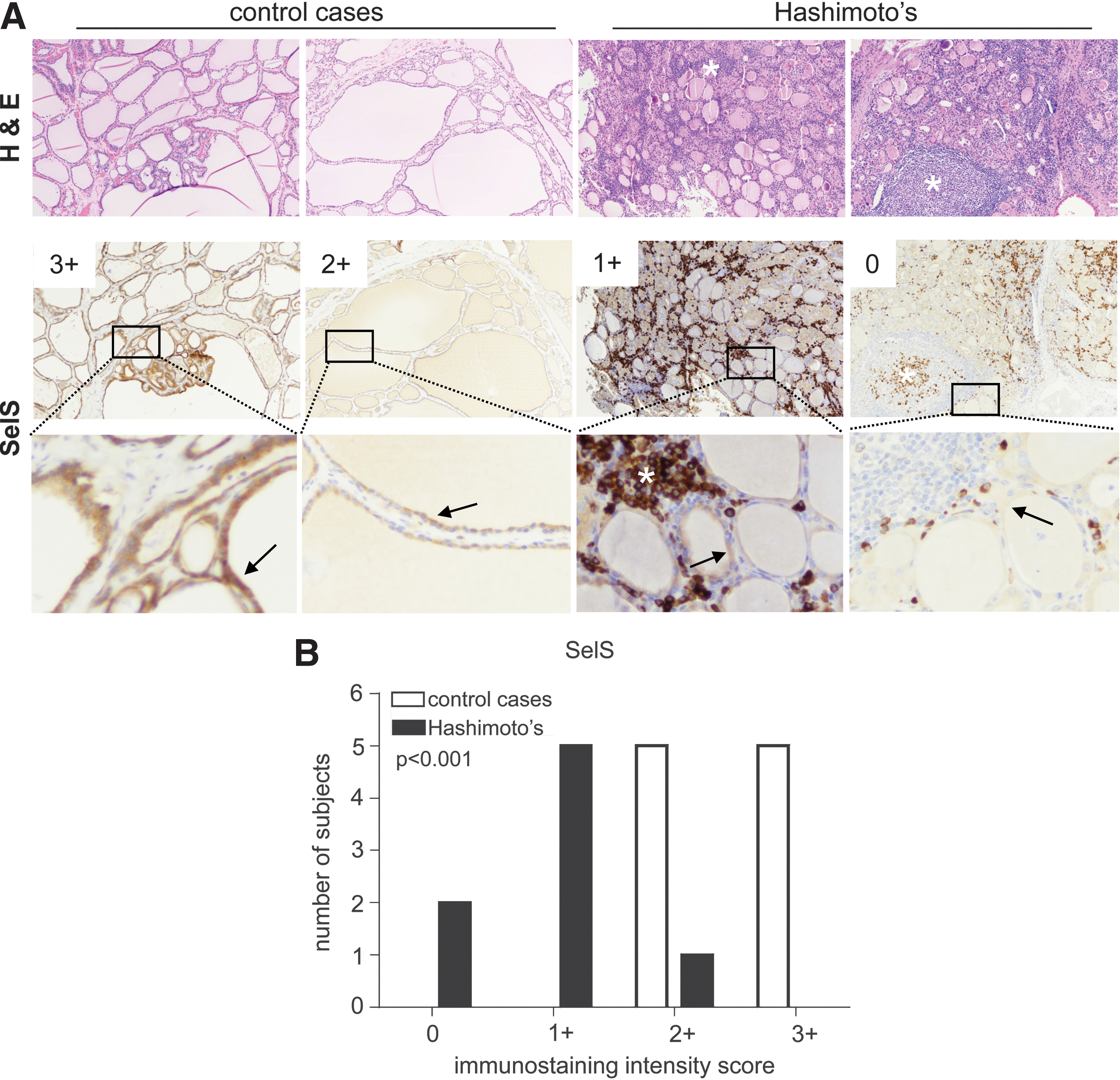
Expression of SelS in samples from control subjects and Hashimoto's patients. (
Reduced expression of SelS in thyroid follicular cells of Nrf2-KO mice
We next analyzed by RT-PCR, Western immunoblotting, and immunohistochemistry the expression of SelS mRNA and SelS protein, respectively, in thyroid samples from WT and Nrf2-KO mice. No difference was found at the SelS mRNA level between the genotypes (Fig. 2A) in thyroid tissue. However, thyroidal SelS protein levels were significantly reduced in Nrf2-KO mice (Fig. 1C, E). Immunohistochemistry showed that this reduction was associated with reduced expression in the thyroid follicular cells (Fig. 1G). When liver tissue samples were analyzed, no differences were observed in either SelS mRNA (Fig. 1B) or SelS protein levels (Fig. 1D, F), suggesting that the observed differences in thyroid are a tissue-specific effect.
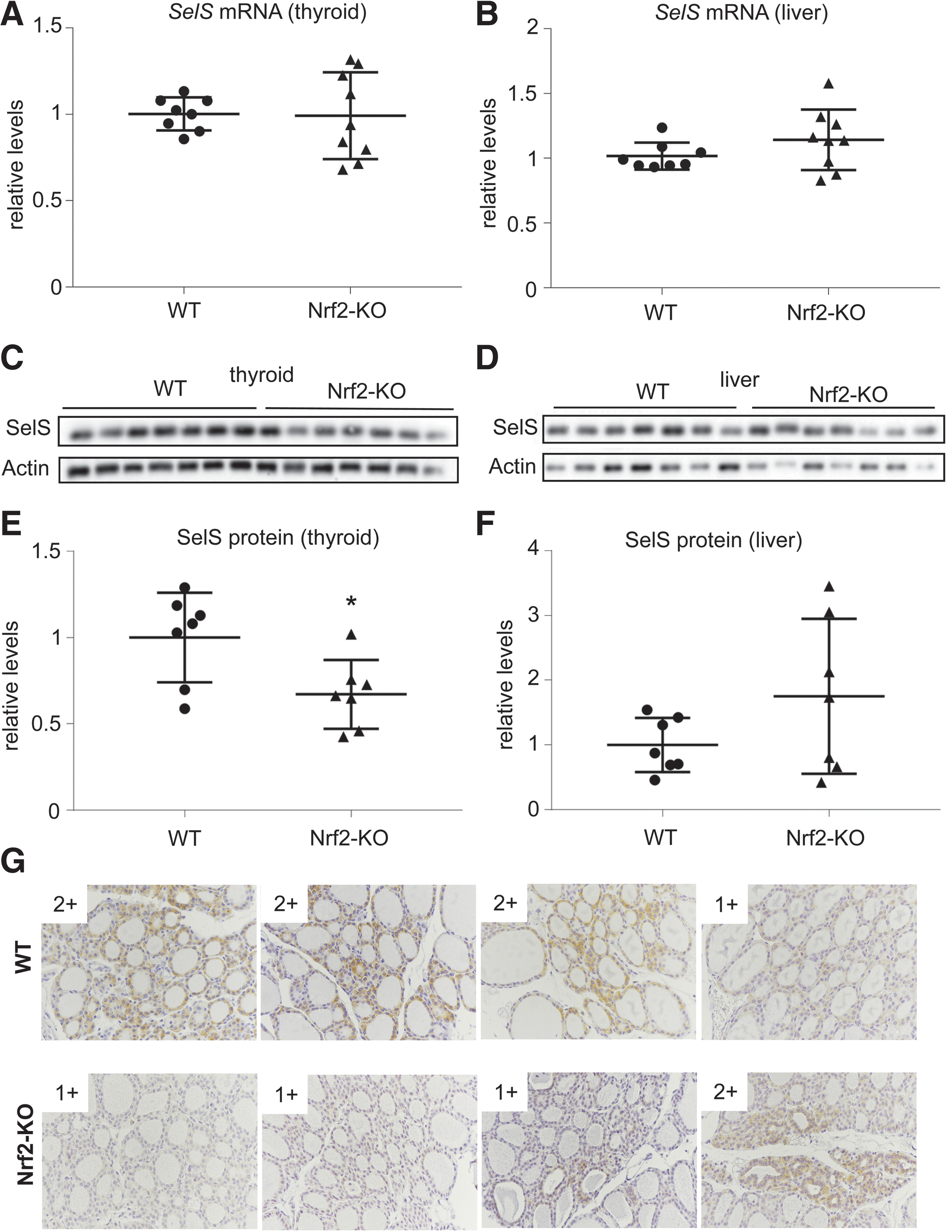
Expression of SelS mRNA and SelS protein in WT and Nrf2-KO mice. (
SELENOS is not a transcriptional target gene of Nrf2
To further investigate how Nrf2 impacts SelS levels, we examined whether it may positively regulate the activity of the human SELENOS promoter. To that end, we cloned three fragments of the SELENOS promoter into luciferase reporter constructs to perform activity assays in transiently transfected PCCL3 rat thyroid follicular cells. All fragments contain a known ER stress-responsive element (52), and the two longer fragments also contain, respectively, one or two predicted ARE sites (Fig. 3A). In both PCCL3 WT and Nrf2-KO cells, all reporter constructs showed significant inducibility by the ER stress-inducing compound TN, but no inducibility in response to treatment with the Nrf2 inducer SLF (Fig. 3B, C). In the same cells, TN, but not SLF, significantly induced the SelS mRNA and SelS protein levels (Fig. 1D, E). Genetic activation of Nrf2 (as observed in Keap1-KO PCCL3 cells (38)) also had no effect on basal or inducible SelS mRNA and SelS protein levels (Fig. 1D, E). Taken together, these data indicate that Nrf2 does not have a direct effect on the SELENOS promoter but that it positively regulates SelS protein levels in thyroid follicular cells in a posttranscriptional manner.
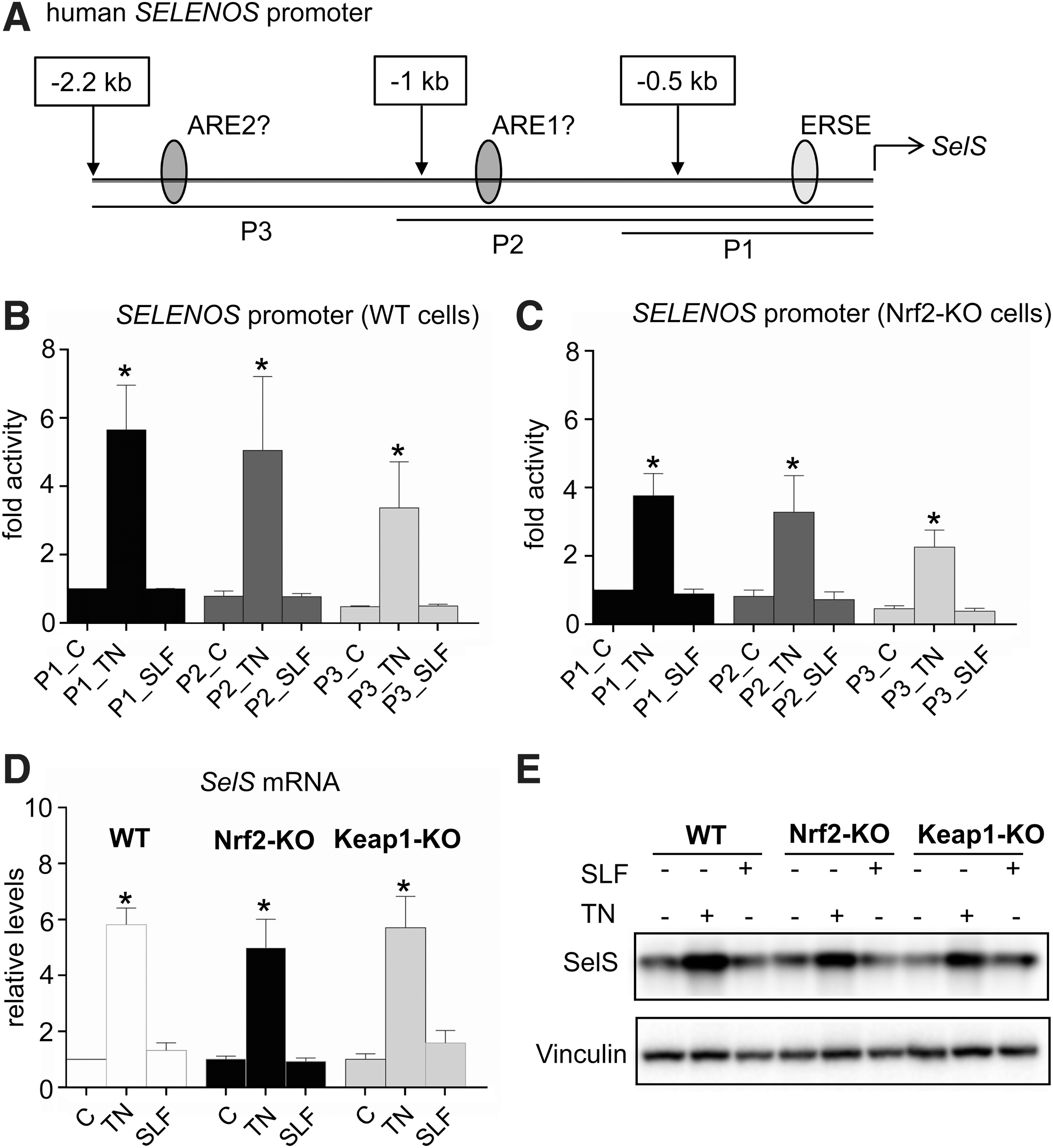
Functional assessment of potential Nrf2-binding sites in the human SELENOS promoter. (
Reduced activity of Nrf2 signaling in thyroid follicular cells with reduced SelS expression
We also aimed to investigate the effects of SelS on Nrf2 signaling. To that end, we first established conditions for an efficient knockdown of SelS in PCCL3 cells, by combining three siRNA constructs yielding a strong additive effect (Fig. 3A, B). SelS knockdown decreased the mRNA expression levels of Nqo1, the prototypical Nrf2 target gene, both under basal conditions and in response to pharmacological induction of Nrf2 by SLF or genetic activation of Nrf2 (Keap1-KO) (Fig. 4C). The thyroidal mRNA levels of Gpx2 and Txnrd1 have also been previously shown to be regulated, in part, by Nrf2 (38); SelS knockdown decreased their mRNA expression levels under basal conditions and in response to genetic activation of Nrf2 (Keap1-KO), but not in response to pharmacological induction of Nrf2 by SLF (Fig. 4D–E), which may be because the latter compound can also modulate other signaling pathways and biological processes (53). Finally, we examined the effects of SelS and Nrf2 on cell viability under conditions of ER stress or oxidative stress. Cell viability was not reduced by treatment with either TN or SLF (Fig. 4F). However, knockdown of SelS reduced cell viability specifically in response to TN treatment, and pretreatment of the cells with SLF was unable to rescue this effect. Conversely, when cells were exposed to oxidative stress generated by treatment with menadione, cells were almost completely unviable, yet SLF pretreatment was able to fully rescue this toxicity. However, when SelS was knocked down, the ability of SLF pretreatment to rescue the viability of cells was significantly reduced. Taken together, these data indicate that SelS positively impacts the activity of Nrf2 signaling and its downstream protective effects against oxidative stress-induced cell toxicity.
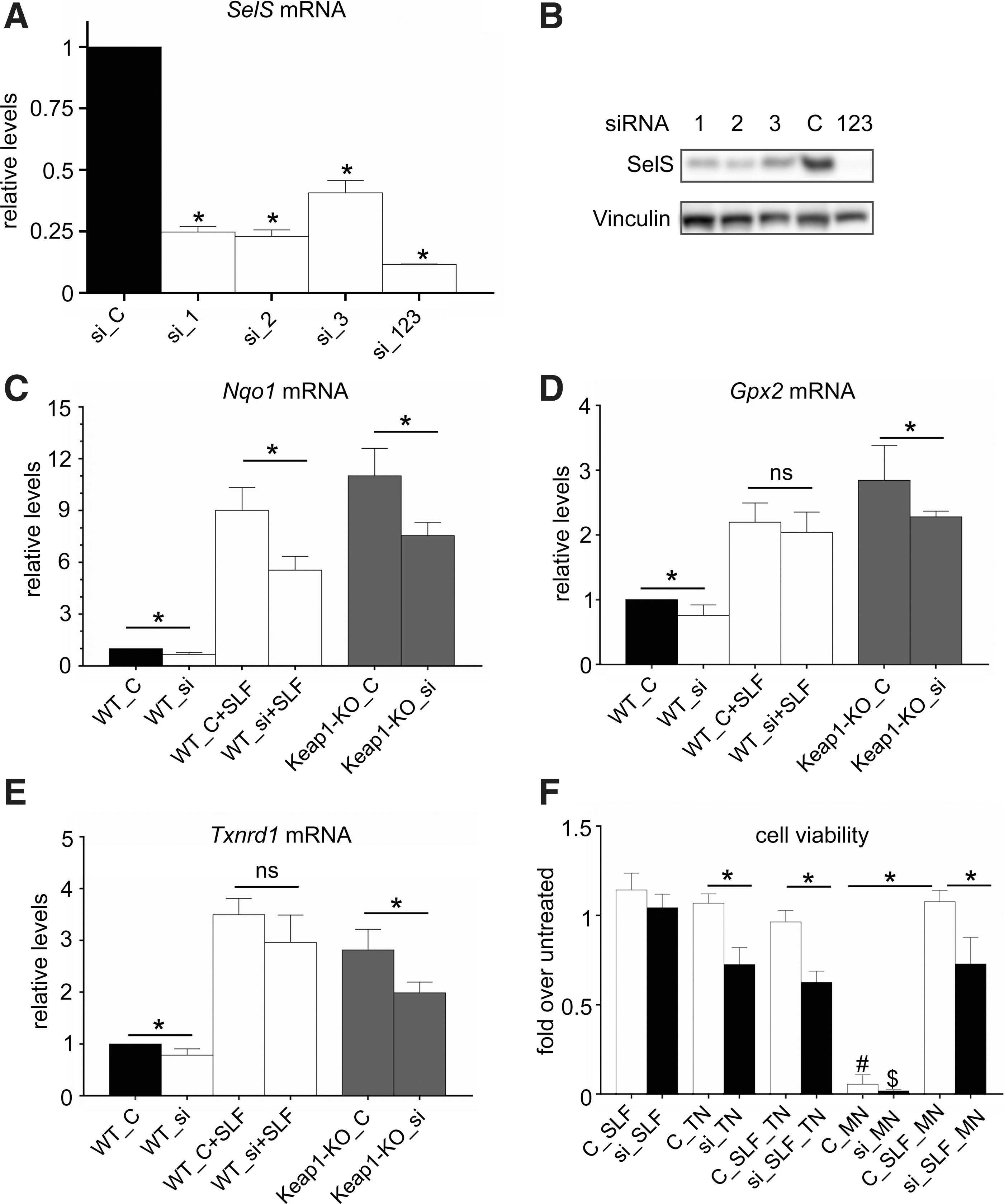
Impact of SelS gene silencing on Nrf2 target gene expression and viability after ER or oxidative stress challenge. (
Discussion
The present study expands the known roles of Nrf2 in the thyroid to include protection from autoimmune thyroid disease. The data indicate that NFE2L2 SNPs do not act in isolation from other genes, but that they interact as a group specifically with the SNP in SELENOS that has been previously associated with HT (10). This is indicated by the results shown in Table 7, which demonstrate not only that NFE2L2 minor alleles confer a risk when present together with the SELENOS minor allele but also that the presence of only NFE2L2 major alleles abolishes the HT risk associated with the SELENOS minor allele. These data are consistent with the signaling interactions between the Nrf2 and SelS systems in thyroid cells described in Figures 2–4, which suggest that there is a certain level of physiological compensation and bidirectional positive feedback between the two pathways.
Previous studies have implicated Nrf2 in papillary thyroid carcinoma (44), goiter (54,55), antioxidant defense in response to excess iodide (38,56), and Tg economy (38). While the mechanism through which the NFE2L2 minor alleles contribute to the risk of HT remains to be characterized, some plausible hypotheses can already be proposed. Using Nrf2 KO mice and thyrocyte cell lines, we have recently shown that Nrf2 mediates antioxidant transcriptional responses in thyroid follicular cells and protects the thyroid from oxidation induced by iodide overload (38). In that work, we also found that Nrf2 has a dramatic impact on both the basal abundance and the TSH-inducible intrathyroidal abundance of Tg, an effect mediated by cell autonomous regulation of TG gene expression by Nrf2 via its direct binding to two evolutionarily conserved AREs in an upstream enhancer (38). Yet, despite upregulating Tg levels, Nrf2 was found to limit Tg iodination both under basal conditions and in response to excess iodide (38). Thus, it is possible that when Nrf2 levels are reduced due to the presence of NFE2L2 minor alleles, Tg iodination is increased. Because increased and aberrant Tg iodination can uncover cryptic antigenic epitopes (57), an increase in Tg iodination associated with NFE2L2 minor alleles might, over the long term, contribute to the pathogenesis of HT. Another possibility concerns the general antiautoimmunity role of Nrf2; this has been demonstrated in studies in Nrf2 KO mice. When these mice are bred into autoimmunity-permissive genetic backgrounds, a subset of them develops a multiorgan autoimmune syndrome (35 –37). These studies indicate that Nrf2 has a protective role against autoimmunity, either via actions in each individual tissue (as Nrf2 is ubiquitously expressed) or via actions in the immune system that impact self-tolerance toward various autoantigens. Of course, the two aforementioned hypotheses (increased Tg iodination and deregulated autoimmunity) are not mutually exclusive.
The present study also expands the known interactions between Nrf2 and the selenium system. Selenium and Nrf2 regulate similar proteostatic processes, including antioxidant defense and the ER stress response; for example, both SelS and Nrf2 function as mediators of ERAD (11,58). Moreover, selenium and Nrf2 regulate proteostasis not only independently but also via converging on common genes and proteins. For example, Nrf2 directly upregulates the transcription of genes encoding selenoproteins, including Gpx2 and Txnrd1 (38), which are both known to participate in redox signaling and antioxidant defense, as well as to have important homeostatic roles in thyroid follicular cells (39,40). In this context, it is not surprising that multiple lines of evidence indicate that the selenoprotein system and the Nrf2 system can compensate for each other in antioxidant stress responses (18,19). For example, moderate selenium deficiency activates Nrf2 signaling in the liver and intestine of mice (20,21). Tissue-specific deletion of the selenocysteine tRNA gene, which inserts selenocysteine into selenoprotein enzymes, results in compensatory induction by Nrf2 of genes encoding cytoprotective enzymes (22). Thus, in these settings, the simultaneous disruption of the two systems is particularly deleterious to proteostasis, leading to enhanced disease phenotypes (22,23). These types of interactions have been documented in animal studies focusing on different tissues, including erythrocyte precursors, macrophages, and liver (22,23). Our present findings using human genetics and complementary human-, animal-, and cell-based studies indicate that such an interaction is also at play in the thyroid gland, which makes sense given that both selenium and oxidation are central elements in thyroid physiology.
The genetic and signaling interactions between the Nrf2 and Sels pathways and their impact on susceptibility or resistance to HT are summarized in the model proposed in Figure 5. Nrf2 and SelS are known to favor resistance to oxidative stress and ER stress, respectively; moreover, both Nrf2 and SelS are also both known to suppress the expression of proinflammatory cytokines, including IL-6 and IL-1β, which are two cytokines known to be involved in the pathogenesis of HT (12,59). In addition to general interactions between the Nrf2 and SelS protective systems, thyroid-specific mechanisms may also exist. For example, one explanation for the lower levels of SelS in the thyroid of Nrf2-KO mice may be the fact that these mice have lower levels of Tg (38). Since Tg comprises the bulk of the thyroid's protein content and represents a high-molecular-weight protein that depends critically on the ER for its proper conformation, the lower Tg levels in Nrf2-KO mice may impose a lower burden on the ER. Previous studies have shown that SelS is positively regulated by ER stress (52), suggesting that lower ER stress levels in Nrf2-KO mice (and, potentially, in carriers of NFE2L2 minor SNP alleles) may lead to decreased SelS expression. Conversely, our findings show that SelS has a positive effect on the activity of Nrf2 signaling, because reduction of SelS levels causes a reduction in the mRNA expression levels of Nrf2 target genes. Interestingly, this effect is also observed in Keap1-KO cells, suggesting that the underlying mechanism is Keap1 independent and thus likely involves a downstream step in Nrf2 signaling. In summary, the presence of a bidirectional positive feedback between the SelS and Nrf2 systems may explain why even small variations in gene expression (as a result of the SNPs) and/or activity of either pathway may be amplified, leading to phenotypic effects. Clearly, the roles of Nrf2 in the thyroid and its cross talk with other proteostatic systems in this gland warrant further elucidation.
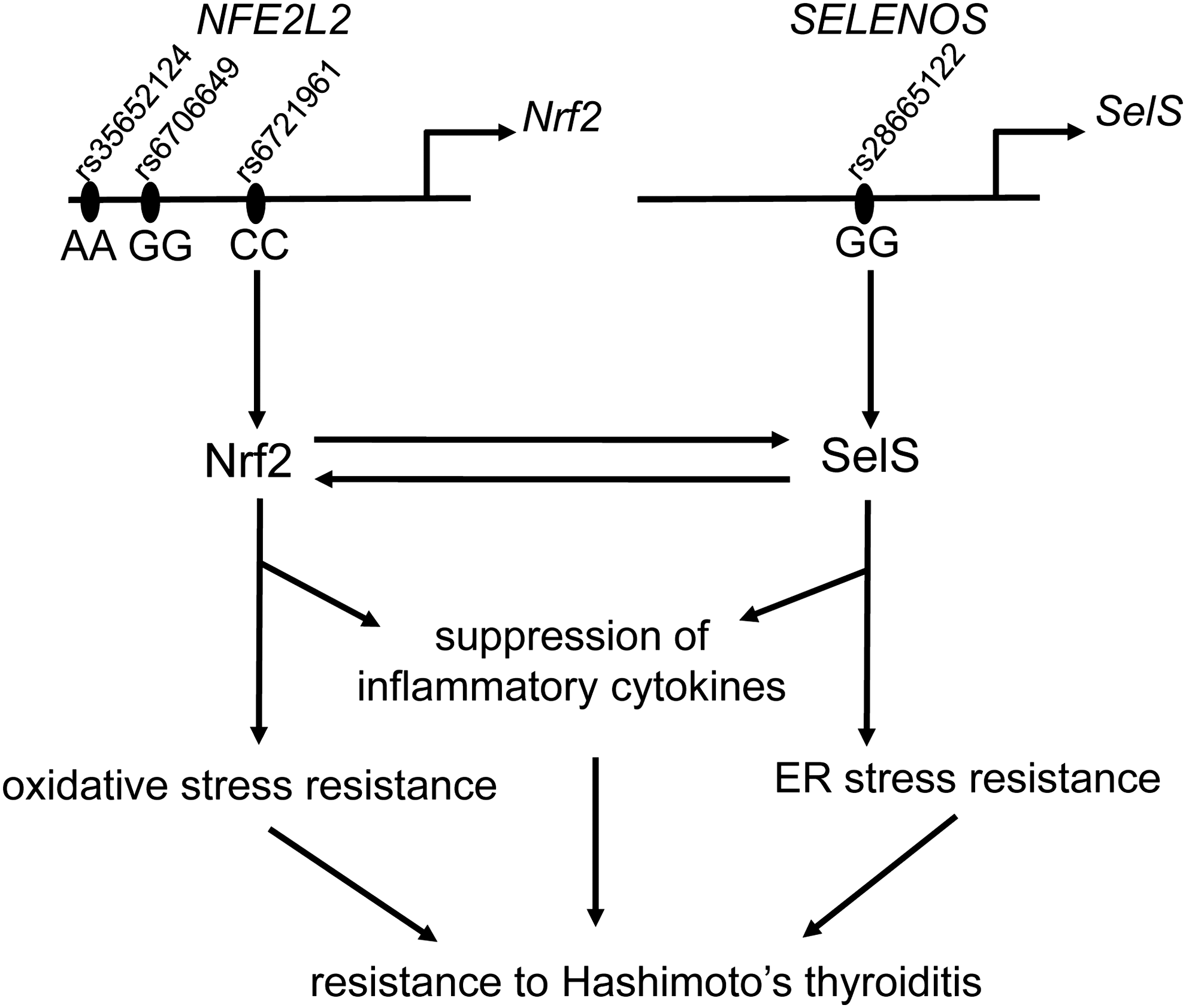
Proposed model of genetic and signaling interactions between the Nrf2 and SelS pathways and their impact on susceptibility or resistance to HT. The model illustrates the expected situation in an individual who is homozygous for the major alleles of the three functional SNPs in NFE2L2 (rs35652124, −214A>G; rs6706649, −212G>A; and rs6721961, −178A>C) and also homozygous for the major allele of the functional SNP in SELENOS (rs28665122, −105G/A). In this setting, the Nrf2 and SelS pathways protect against the development of HT, possibly by suppressing the expression of inflammatory cytokines and promoting resistance to oxidative and ER stress. The present study also provides evidence of a bidirectional positive feedback between the Nrf2 and SelS pathways. Specifically, Nrf2 positively regulates SelS protein levels in thyroid follicular cells in a posttranscriptional manner. Conversely, SelS positively impacts the activity of Nrf2 signaling and its downstream protective effects against oxidative stress-induced cell toxicity. HT, Hashimoto's thyroiditis.
This study has some limitations. First, it is acknowledged that a biological interaction between the two pathways is not an independent proof of a genetic interaction (epistasis). Second, it is interesting that subjects with additional NFE2L2 SNP minor alleles (from 2 to 6 in total) had the same level of risk for developing HT as those with a single minor allele. Even though a simple gene dosage model is easier to conceptualize, for a multifactorial disease of essentially unknown pathogenesis such as HT, other models, such a threshold model, are equally plausible. In fact, we speculate that a signaling interaction between the two genetically interacting pathways might actually better fit a threshold model. Finally, it is acknowledged that the observed interaction may not apply to all ethnicities. A recent genome-wide association study (GWAS) performed on a Croatian population suggested novel loci for HT, but did not find associations with SELENOS or NFE2L2 variants (60). Of note, that study did not test specifically for an interaction between SELENOS and NFE2L2 variants. Also, there were several factors that differentiate it from the present study, including the very specific and distinct ethnic origins of the participants in the two studies (Portuguese vs. Croatian); the different clinical profiles of HT patients in the two studies (e.g., overt hypothyroidism in all HT patients in the Portuguese cohort vs. inclusion of HT patients with subclinical hypothyroidism or even spontaneous euthyroidism in the Croatian study); and the relatively small population size (for a GWAS) of the Croatian cohort (838 participants with usable data in the discovery cohort) (60). The presence of striking ethnic patterns in HT susceptibility indicating the implication of ethnically restricted effects has been well acknowledged (3). It will thus be interesting to perform replication studies for the genetic interaction between SELENOS and NFE2L2 variants in various independent ethnic cohorts of HT patients.
The importance of selenium for the thyroid has long been known. Selenium supplementation is recommended for the treatment of orbitopathy associated with autoimmune hyperthyroidism (Graves' disease) (61), and it is also currently being tested in a randomized controlled clinical trial as a therapeutic measure for chronic autoimmune thyroiditis (HT) (62). In view of the present findings, it might be interesting to test combinations of selenium with Nrf2 activating compounds, such as SLF-containing natural extracts, whose safety for the thyroid has actually been shown in a recent clinical trial (63).
Footnotes
Author Disclosure Statement
The authors have nothing to disclose.
Funding Information
This study was supported by an IPG-UP grant (with financial support from Caixa Geral de Depósitos) to LRS; FCT, the Portuguese Foundation for Science and Technology through a PhD grant to AP SFRH/BD/110617/2015; FCT Postdoc grant SFRH/BPD/99442/2014 to CD; FEDER—Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020—Operational Programme for Competitiveness and Internationalization (POCI), Portugal 2020; through FCT in the framework of the project “Institute for Research and Innovation in Health Sciences” (POCI-01-0145-FEDER-007274); “Advancing cancer research: from basic knowledge to application”; NORTE-01-0145-FEDER-000029; and “Projetos Estruturados de I&D&I,” funded by Norte 2020-Programa Operacional Regional do Norte to PS; FP7-PEOPLE-2009-RG268266 to GPS; SSED/SGED Young Independent Investigator Award 2014 to GPS; Swiss National Science Foundation grants 31003A_153062-1 and 31003A_182105/1 to GPS, Swiss State Secretariat for Education, Research and Innovation COST—Swiss National Science Foundation Research Projects No. C15.0045-174626 and IZCOZ0_177070/1 to GPS, 3R Foundation Switzerland Project Grant No. 146–15 to GPS, and Leenaards Foundation 2016 Fellowship for Academic Promotion in Clinical Medicine to GPS.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3