
Abstract
Background:
Thyroid hormones act in bone and cartilage via thyroid hormone receptor alpha (TRα). In the absence of triiodothyronine (T3), TRα interacts with co-repressors, including nuclear receptor co-repressor-1 (NCoR1), which recruit histone deacetylases (HDACs) and mediate transcriptional repression. Dominant-negative mutations of TRα cause resistance to thyroid hormone alpha (RTHα; OMIM 614450), characterized by excessive repression of T3 target genes leading to delayed skeletal development, growth retardation, and bone dysplasia. Treatment with thyroxine has been of limited benefit, even in mildly affected individuals, and there is a need for new therapeutic strategies. It was hypothesized that (i) the skeletal manifestations of RTHα are mediated by the persistent TRα/NCoR1/HDAC repressor complex containing mutant TRα, and (ii) treatment with the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) would ameliorate these manifestations.
Methods:
The skeletal phenotypes of (i) Thra1PV/+ mice, a well characterized model of RTHα; (ii) Ncor1ΔID/ΔID mice, which express an NCoR1 mutant that fails to interact with TRα; and (iii) Thra1PV/+Ncor1ΔID/ΔID double-mutant adult mice were determined. Wild-type, Thra1PV/+ , Ncor1ΔID/ΔID , and Thra1PV/+Ncor1ΔID/ΔID double-mutant mice were also treated with SAHA to determine whether HDAC inhibition results in amelioration of skeletal abnormalities.
Results: Thra1PV/+
mice had a severe skeletal dysplasia, characterized by short stature, abnormal bone morphology, and increased bone mineral content. Despite normal bone length, Ncor1ΔID/ΔID mice displayed increased cortical bone mass, mineralization, and strength. Thra1PV/+Ncor1ΔID/ΔID double-mutant mice displayed only a small improvement of skeletal abnormalities compared to Thra1PV/+ mice. Treatment with SAHA to inhibit histone deacetylation had no beneficial or detrimental effects on bone structure, mineralization, or strength in wild-type or mutant mice.
Conclusions:
These studies indicate treatment with SAHA is unlikely to improve the skeletal manifestations of RTHα. Nevertheless, the findings (i) confirm that TRα1 has a critical role in the regulation of skeletal development and adult bone mass, (ii) suggest a physiological role for alternative co-repressors that interact with TR in skeletal cells, and (iii) demonstrate a novel role for NCoR1 in the regulation of adult bone mass and strength.
Introduction
Triiodothyronine (T3) acts mainly via thyroid hormone receptor alpha (TRα) in bone and cartilage and regulates skeletal development, postnatal growth, and the maintenance of adult bone mass, mineralization, and strength (1). Mutations affecting THRA encoding TRα cause resistance to thyroid hormone alpha (RTHα) (2 –12), also classified as autosomal dominant non-goitrous congenital hypothyroidism type 6 (OMIM 614450).
THRA mutations result in a consistent pattern of thyroid function test abnormalities, comprising normal levels of thyrotropin (TSH), a low or normal thyroxine (T4), and a high or normal T3 concentration. An elevated T3/T4 ratio is pathognomonic and present in all affected individuals (10). Individuals with RTHα display a characteristic skeletal dysplasia consistent with impaired T3 action in bone and the skeletal consequences of severe congenital or juvenile hypothyroidism (1,10). These include macrocephaly with patent fontanelles and cranial sutures, delayed tooth eruption, thickened calvarium with wormian sutures, delayed ossification and bone age, epiphyseal dysgenesis, and disproportionate short stature. Affected adults have cortical hyperostosis and increased bone mineral density (BMD). A phenotype–genotype correlation has been noted in the limited number of reported cases. Missense mutations are associated with a less severe phenotype than the profound dysplasia in individuals with truncation mutations that result in expression of potent dominant-negative mutant TRα proteins (1,7,10). Consistent with this, the degree of dominant-negative activity of mutant TRα also correlates with the clinical response to treatment with thyroid hormones (6,10).
In RTHα, the mutant TRα acts as a dominant-negative repressor of T3 target gene expression and an inhibitor of wild-type TR function (2). In the absence of T3, unliganded TRα and TRβ isoforms interact with transcriptional repressors, including nuclear receptor co-repressor-1 (NCoR1). This interaction leads to recruitment of histone deacetylase (HDAC) enzymes to a co-repressor complex, resulting in chromatin remodeling and inhibition of basal T3 target gene transcription (13). Binding of T3 causes a conformational change in the receptor and disrupts the interaction between TR and NCoR1. T3 binding thus promotes recruitment of nuclear receptor co-activators, such as steroid receptor co-activator 1, which possess histone acetyl transferase activity, leading to activation of T3 target gene expression (14). In RTHα, the mutant TRα protein cannot release NCoR1 in response to T3, resulting in dominant repression of T3 target gene transcription because persistent HDAC-induced chromatin remodeling also prevents access for wild-type TRs to the transcriptional machinery. The disease phenotype in RTHα therefore reflects impaired T3 action in specific TRα-dependent target tissues such as the skeleton, and its severity is directly related to the dominant-negative potency of the mutant receptor.
It was hypothesized that (i) the skeletal manifestations of RTHα are mediated by the persistent TRα/NCoR1/HDAC repressor complex containing mutant TRα, and (ii) treatment with the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) would ameliorate the skeletal abnormalities.
To investigate these hypotheses, Thra1PV/+ mice, which express a potent dominant negative mutant TR (TRα1PV) and recapitulate the RTHα phenotype observed in individuals with similar THRA mutations, were studied (15 –17). To determine the role of NCoR1 in the pathogenesis of the skeletal manifestations of RTHα, Thra1PV/+ mice were crossed with Ncor1ΔID/ΔID mice that express mutant NCoR1, which lacks the receptor interacting domains RID2 and RID3 required for TR binding but retains RID1 that interacts with other nuclear receptors (13,18,19). Finally, wild-type, Thra1PV/+ , Ncor1ΔID/ΔID , and Thra1PV/+Ncor1ΔID/ΔID double-mutant mice were treated with SAHA to determine whether HDAC inhibition results in amelioration of skeletal abnormalities in RTHα.
Methods
Animals and treatment
Animal studies were performed according to protocols approved by the National Cancer Institute Animal Care and Use Committee. Heterozygous Thra1PV/+ mice were generated in a mixed C57BL/6J and NIH Black Swiss genetic background and genotyped as described (17). Homozygous Ncor1ΔID/ΔID mice were generated in a mixed C57BL/6 and 129S6 background as described (20). Thra1PV/+ mice and Ncor1ΔID/ΔID mice were inter-crossed for several generations to produce Thra1PV/+Ncor1ΔID/ΔID double mutants in a mixed C57BL/6J, NIH Black Swiss and 129S6 genetic background (21). Wild-type mice in a mixed C57BL/6J, NIH Black Swiss and 129S6 genetic background were used to ensure comparisons were made in as similar a genetic background as possible for all experiments (n = 4–8 per group).
SAHA (Selleck Chemicals, Houston, TX) or vehicle was prepared as described (22). A daily dose of 50 mg/kg body weight was administered by oral gavage for a two-month period starting at the age of six weeks until bones were harvested at 14 weeks of age.
Faxitron digital X-ray microradiography
Dissected femurs, humeri, and proximal caudal vertebrae Ca6 and Ca7 were imaged at 10 μm resolution using a Faxitron MX20 (Qados; Cross Technologies plc, Sandhurst, United Kingdom). Bone lengths were determined after calibrating images with a digital micrometer using ImageJ (
Micro computed tomography
Femurs were imaged by micro computed tomography using a SCANCO μCT 50 (SCANCO Medical AG, Bruttisellen, Switzerland) at 70 kV, 200 μA, with a 0.5 mm aluminum filter and voxel resolutions of 5 and 10 μm for trabecular and cortical bone, respectively. Images were reconstructed and analyzed using Scanco software. A 1 mm3 region of interest was selected 100 μm from the growth plate, and trabecular bone volume as proportion of tissue volume (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), and trabecular spacing (Tb.Sp) were determined (23,25,26). Total cross-sectional area (Tt.Ar), cortical bone area (Ct.Ar), marrow or medullary area (Ma.Ar), and cortical area fraction (Ct.Ar/Tt.Ar) were also determined. A 1.5 mm region of interest, centered in the midshaft 56% along the length of the femur distal to the femoral head, was selected to determine cortical thickness (Ct.Th) and cortical BMD. Cortical porosity (Ct.Po), periosteal perimeter (Ps.Pm), and endocortical perimeter (Ec.Pm) were determined within a 0.5 mm region of interest centered in the midshaft 56% along the length of the femur distal to the femoral head from images with a voxel resolution of 1 μm.
Biomechanical testing
Destructive three-point bend tests were performed on 70% ethanol-fixed humeri to determine bone strength using an Instron 5543 load frame and 100 N load cell (Instron Limited, High Wycombe, United Kingdom). Humeri were positioned horizontally on custom supports, and load was applied perpendicular to the mid-diaphysis at a constant rate of displacement of 0.03 mm/s until fracture. Yield load, maximum load, fracture load, and stiffness were determined from load displacement curves (23,27).
Statistical analysis
Data were normally distributed and analyzed by analysis of variance and Tukey's post hoc test or unpaired two-tailed Student's t-test. p-Values <0.05 were considered statistically significant. Cumulative and relative frequency distributions of BMC were compared using the Kolmogorov–Smirnov test (23,24).
Results
Expression of NCoR1ΔID ameliorates bone structural defects in Thra1PV/+ mice and increases bone strength
In 14-week-old adult Thra1PV/+ mice, the lengths of the femurs, humeri, and vertebrae were decreased by 12%, 16%, and 30%, respectively, and BMC was increased (p < 0.001). The changes in bone length and BMC were accompanied by morphological abnormalities that included dysmorphic epiphyses with misshapen joints and splayed metaphyses with defective inwasting (Fig. 1A). Similar dysplastic features were evident in Thra1PV/+Ncor1ΔID/ΔID double-mutant mice, in which bone lengths were also decreased and BMC increased compared to wild-type mice. Nevertheless, reductions in the lengths of the humerus and vertebrae in Thra1PV/+Ncor1ΔID/ΔID double mutants were less than those observed in Thra1PV/+ mice (Fig. 1A). Femurs from Thra1PV/+ mice had reduced Ct.Th and increased Ct.Po but no difference in BMD compared to wild-type mice (Fig. 1B). These parameters were accompanied by increased Tt.Ar, Ma.Ar, Ps.Pm, and Ec.Pm, no difference in Ct.Ar, and a decrease in Ct.Ar/Tt.Ar (Supplementary Fig. S1), indicating an overall decrease in cortical bone thickness together with increased porosity but normal BMD. Thra1PV/+ mice also had high trabecular bone mass, as evidenced by a threefold increase in BV/TV, a twofold increase in Tb.N, and a twofold reduction in Tb.Sp (Fig. 1C). Thra1PV/+Ncor1ΔID/ΔID double-mutant mice had a small increase in BMD and increased Ct.Po (Fig. 1B) but high trabecular bone mass, with a 2.5-fold increase in BV/TV, a 50% increase in Tb.N, a 10% increase in Tb.Th, and a 62.5% reduction in Tb.Sp (Fig. 1C). The changes in cortical and trabecular bone parameters were not as large in Thra1PV/+Ncor1ΔID/ΔID double mutants compared to Thra1PV/+ mice (Fig. 1B and C), other than an increase in cortical area (Supplementary Fig. S1). The structural abnormalities in Thra1PV/+ mice resulted in humeri that were weak, with an 18% decrease in yield load and 16% decrease in maximum load, whereas humeri from Thra1PV/+Ncor1ΔID/ΔID double-mutant mice were stronger, with a 16% increase in maximum load and 24% increase in fracture load compared to wild-type mice (Fig. 1D).
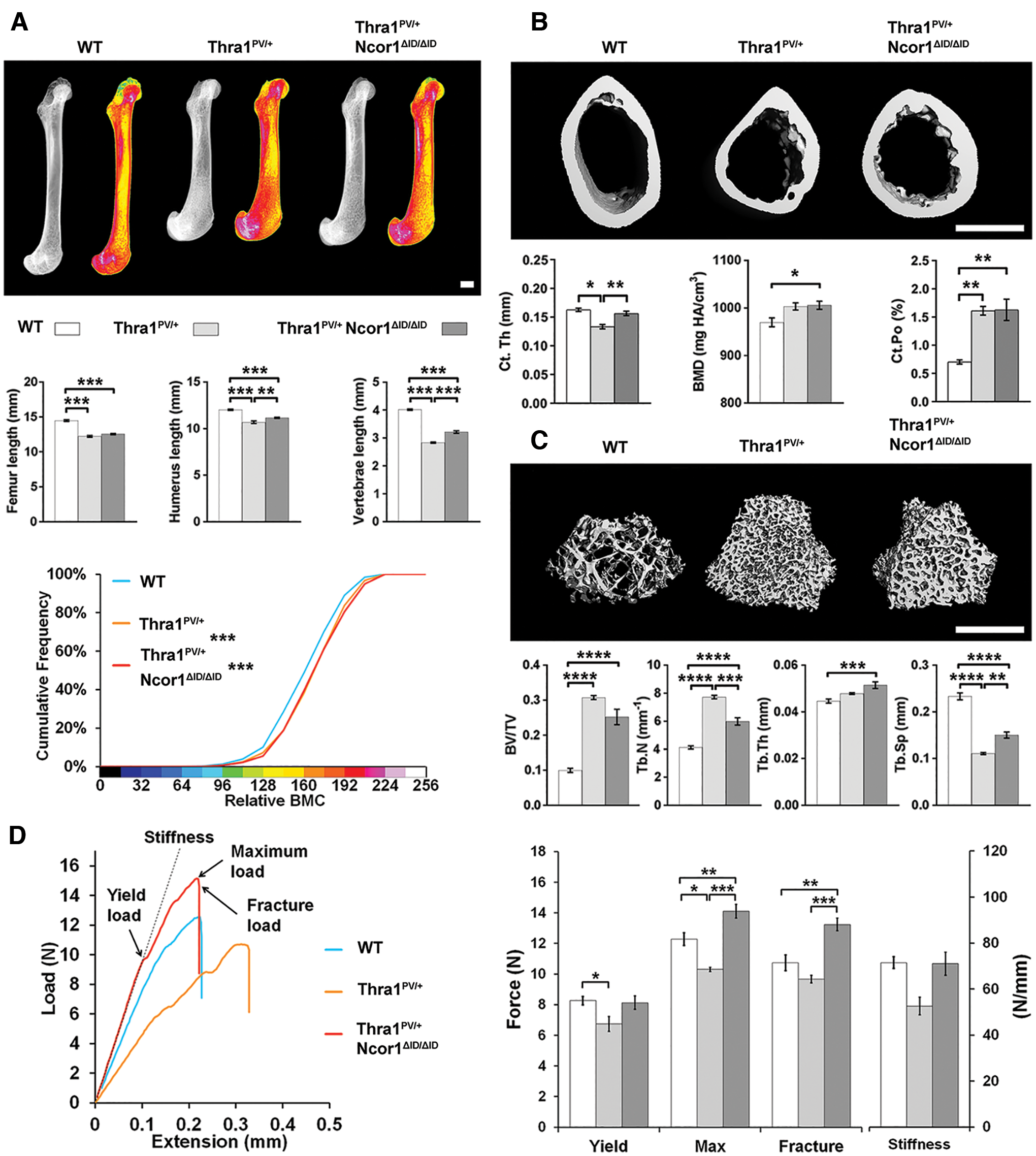
Disruption of the interaction between nuclear receptor co-repressor-1 (NcoR1) and thyroid hormone receptor alpha (TRα) ameliorates the skeletal phenotype in Thra1PV/+
mice. (
Overall, expression of NCoR1ΔID in double-mutant mice ameliorated the structural abnormalities evident in bones from Thra1PV/+ mice and ultimately resulted in increased adult bone strength, consistent with the increased cortical area observed in Thra1PV/+Ncor1ΔID/ΔID double mutants.
Expression of NCoR1ΔID increases bone mass, mineralization, and strength
In 14-week-old adult Ncor1ΔID/ΔID mice, skeletal morphology and the lengths of the femurs, humeri, and vertebrae were similar to wild-type mice, but femurs had increased BMC (Fig. 2A) that resulted from a combination of an 18% increase in Ct.Th, a small increase in cortical BMD, and an increase in Ct.Po (Fig. 2B). There were no differences in trabecular bone parameters (Fig. 2C). The increased cortical bone in Ncor1ΔID/ΔID mice (Fig. 2B and Supplementary Fig. S2) resulted in increased bone strength compared to wild-type mice, as evidenced by a 30% increase in maximum load, a 35% increased fracture load, and a 20% increase in stiffness (Fig. 2D).
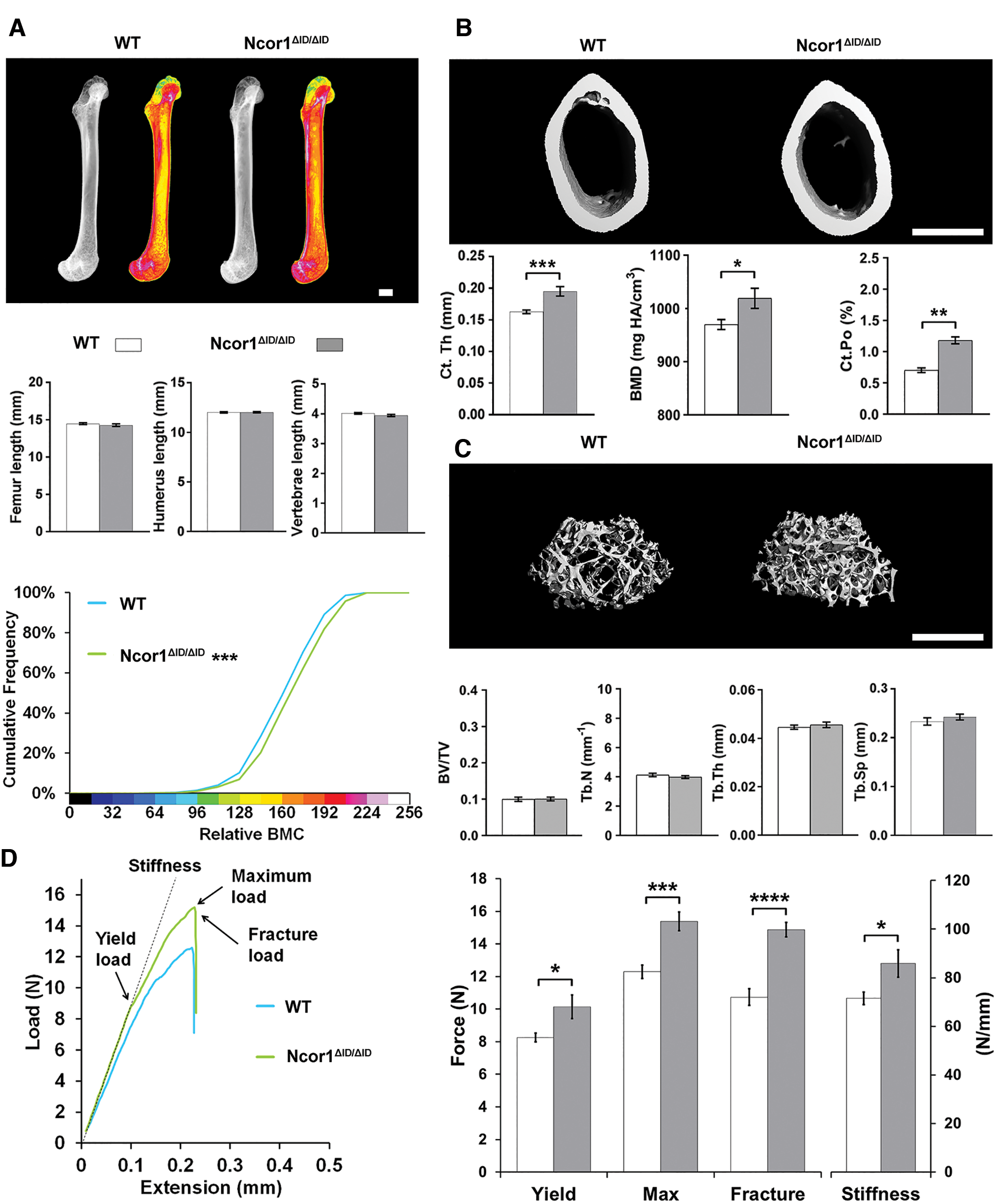
Disruption of the interaction between NcoR1 and TRα increases bone mass, mineralization, and strength in WT mice. (
Overall, expression of NCoR1ΔID results in increased cortical bone mass, mineralization, and strength in adult Ncor1ΔID/ΔID mice despite an increase in Ct.Po.
Treatment with SAHA has no effect on bone mass, mineralization, or strength
Two months of treatment with the histone deacetylase inhibitor SAHA between 6 and 14 weeks of age had no effect on bone structure, mineralization, or strength in either wild-type or Thra1PV/+ mice, other than a small decrease in femur BMC in Thra1PV/+ mice (Fig. 3 and Supplementary Fig. S3). Similarly, treatment of Ncor1ΔID/ΔID and Thra1PV/+Ncor1ΔID/ΔID double-mutant mice with SAHA had no effect on any parameter, other than a small reduction in the length of the humerus present in Thra1PV/+Ncor1ΔID/ΔID double-mutant mice treated with SAHA (Fig. 4 and Supplementary Fig. S4).
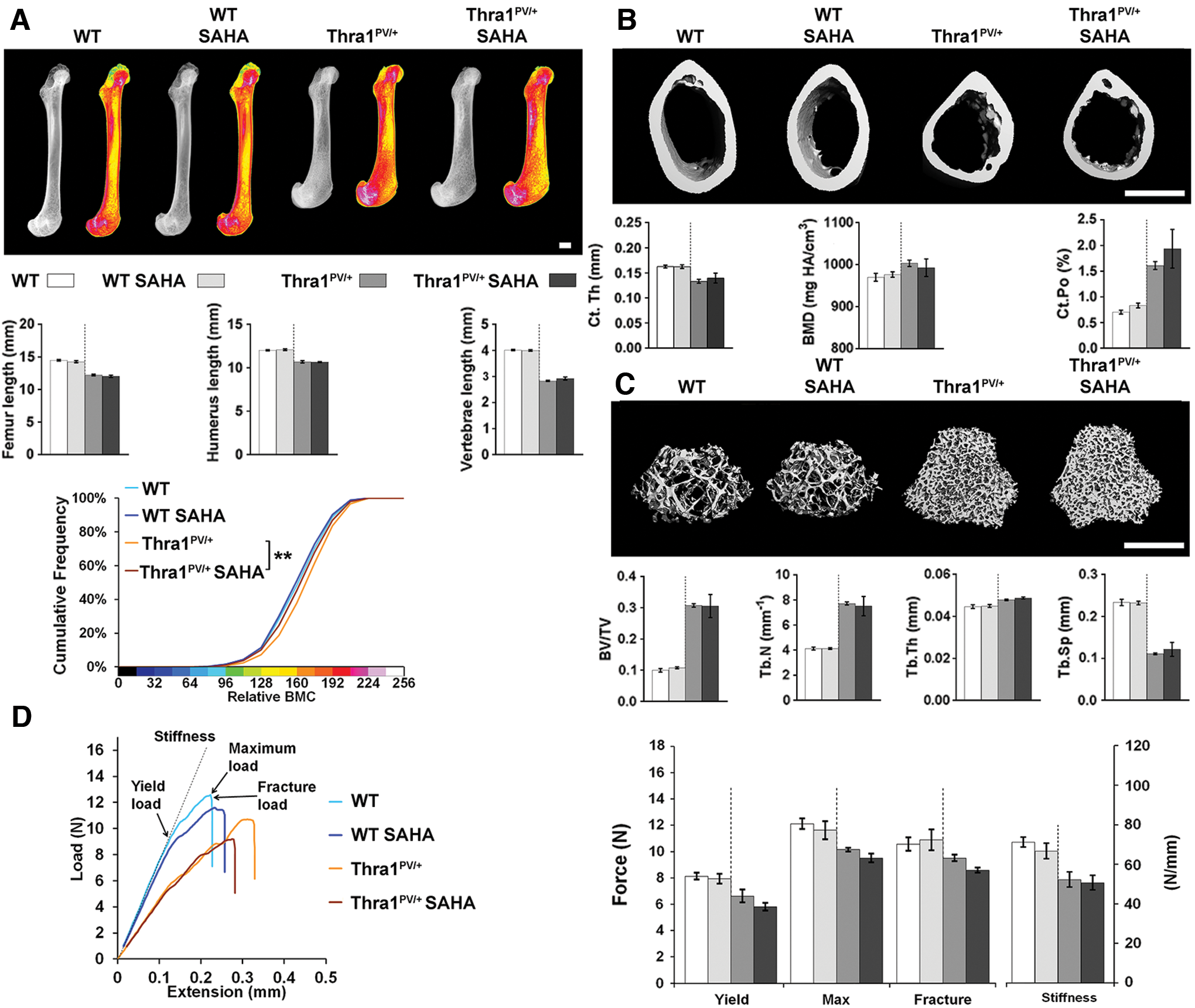
Treatment with suberoylanilide hydroxamic acid (SAHA) has no effect on bone mass, mineralization, or strength in WT or Thra1PV/+
mice. (
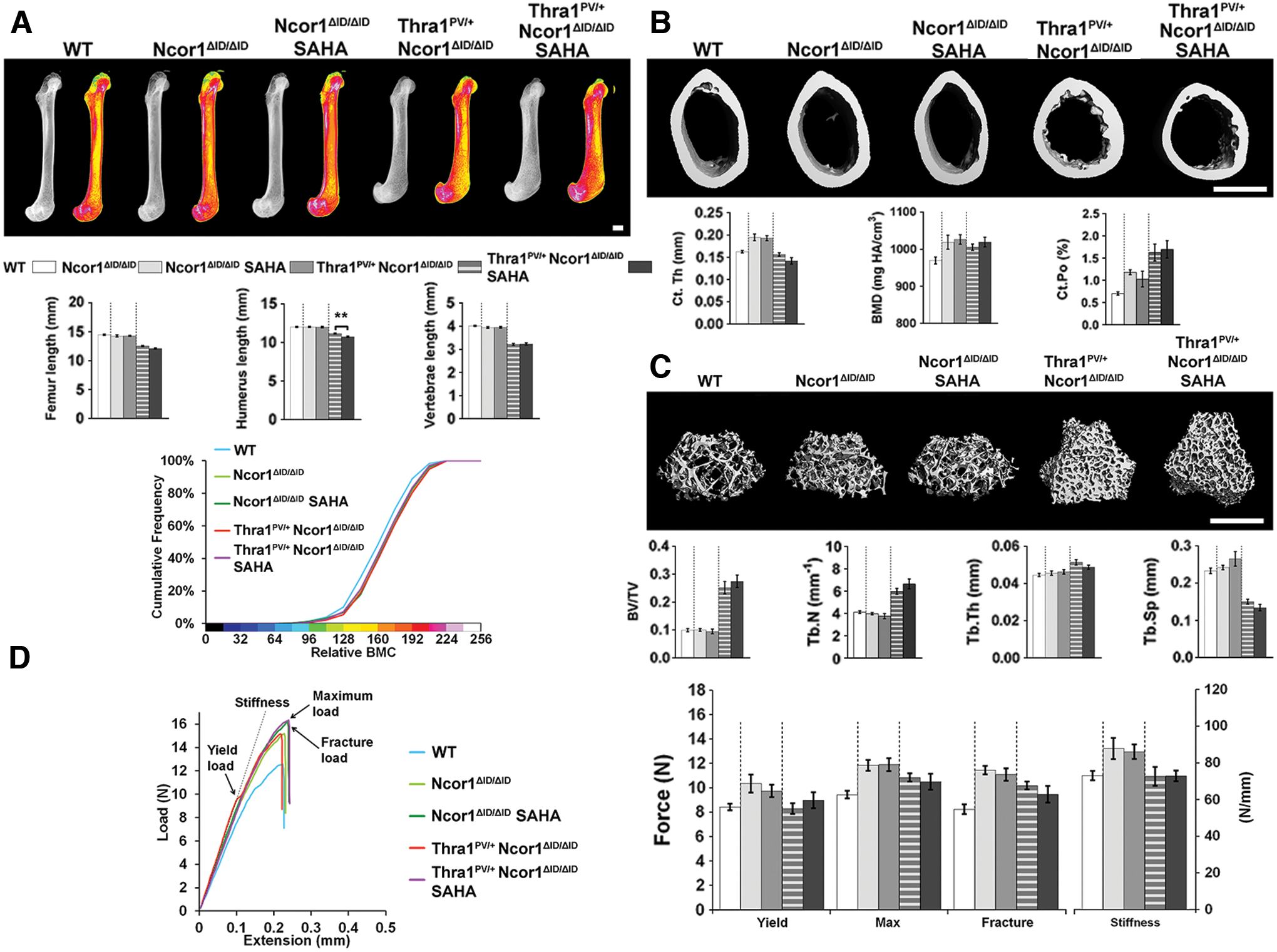
Treatment with SAHA has no effect on bone mass, mineralization, or strength in NCoR1ΔID/ΔID
or Thra1PV/+NCoR1ΔID/ΔID
mice. (
Overall, treatment with SAHA had no beneficial or detrimental effect on bone structure, mineralization, or strength in wild-type or mutant mice.
Discussion
RTHα results in delayed skeletal and dental development, growth retardation, and skeletal dysplasia, as well as additional non-skeletal manifestations primarily affecting the central nervous, gastro-intestinal, and metabolic systems (10). To date, 11 individuals have received treatment with T4, but other therapeutic possibilities have not been investigated (2 –5,8,9,12). Children with less severe frameshift THRA mutations have derived only limited benefit after treatment with T4, particularly with respect to growth (2,8,12), but no skeletal responses have been seen in individuals with deleterious frame-shift truncation mutations (6,9,28). Thus, there is a need to investigate new therapeutic strategies for treatment of the debilitating skeletal manifestations of RTHα. Here, two complementary approaches to disrupt the detrimental dominant-negative activity of mutant TRα in the Thra1PV/+ mouse model of RTHα were investigated.
First, T3 action in bone is mediated by the canonical genomic actions of TRα1 (1,26), while the skeletal dysplasia in RTHα results from (i) impaired tissue T3 responsiveness in bone and cartilage despite a moderately increased circulating T3 concentration and (ii) the dominant-negative repressor activity of mutant TRα1 that results from its inability to release co-repressor in the presence of T3 (13). Normally, NCoR1 interacts with TR via two interaction domains and is the main co-repressor that recruits HDACs to mediate transcriptional repression in the absence of T3. The functional interaction between TRα1 and NCoR1 can be targeted elegantly in mice by the Ncor1ΔID mutation (13,18,19), which prevents association between NCoR1 and unliganded TR and thus relieves NCoR1-dependent transcriptional repression (21,29 –31).
It was previously shown that similar to patients with RTHα, Thra1PV/+ mice have an abnormal hypothalamic–pituitary–thyroid (HPT) axis, with a moderately increased T3 concentration, normal T4, slightly elevated TSH, and an increased size of the thyroid gland (21). Ncor1ΔID/ΔID mice by contrast have decreased T3 and T4 levels but a normal TSH and normal sized thyroid gland. Thra1PV/+Ncor1ΔID/ΔID double-mutant mice have a normal T3, decreased T4, normal TSH, and a slightly enlarged thyroid gland. Thus, co-expression of NCoR1ΔID in Thra1PV/+Ncor1ΔID/ΔID double mutants ameliorates dysregulation of the HPT axis and normalizes the increased T3 and TSH levels that are caused by dominant-negative actions of TRα1PV in Thra1PV/+ mice (21). By contrast, treatment of Thra1PV/+ , Ncor1ΔID/ΔID , and Thra1PV/+Ncor1ΔID/ΔID double-mutant mice with SAHA does not affect circulating T3, T4, or TSH concentrations (22).
Disruption of the interaction between TRα1 and NCoR1 in Ncor1ΔID/ΔID mice resulted in increased cortical bone mass, mineralization, and strength but did not affect linear growth or trabecular bone parameters (Fig. 2). These findings demonstrate a new and important physiological and homeostatic function of NCoR1 affecting the skeleton. Thus, a lack of interaction between NCoR1 and TR isoforms has an anabolic impact on cortical bone to improve bone strength, whereas global interactions between NCoR1 and TRs limit cortical bone accumulation and decrease bone strength. They also reveal that NCoR1 is dispensable during endochondral bone formation and postnatal growth. Consistent with this new role for NCoR1 in bone, disruption of the interaction between dominant-negative TRα1PV and NCoR1 in Thra1PV/+Ncor1ΔID/ΔID double-mutant mice also increased cortical bone mass and strength but had only limited effects on skeletal morphology, linear growth, and trabecular bone mass (Fig. 1). Importantly, comparison of Ncor1ΔID/ΔID mice (Fig. 2) with Thra1PV/+Ncor1ΔID/ΔID double-mutant mice (Fig. 1) reveals that Thra1PV/+Ncor1ΔID/ΔID mice have decreased bone strength parameters. This effect of TRα1PV to worsen the phenotype of Ncor1ΔID/ΔID mice is independent of NCoR1 function and thus provides evidence of a role for additional co-repressors that may interact with TRα in skeletal cells.
Our second approach was to investigate pharmacological inhibition of excessive HDAC activity in RTHα. SAHA chelates zinc ions required for histone deacetylase activity and transcriptional repression, thus resulting in enzyme inhibition, increased histone acetylation, and transcriptional activation (32). SAHA has received approval from the U.S. Food and Drug Administration for the treatment of certain types of cancer and thus could be repurposed for other disease applications if effective. Accordingly, SAHA has been proposed as a potential drug to relieve the detrimental consequences of transcriptional repression in RTHα, and has been shown to ameliorate impaired adipogenesis in Thra1PV/+ mice at the same dose used in the current study (22). Despite this, it was found that pharmacological inhibition of excessive HDAC activity in Thra1PV/+ and Thra1PV/+Ncor1ΔID/ΔID double-mutant mice had no beneficial effect on linear growth or adult bone structure and strength (Figs. 3 and 4). This lack of skeletal response to SAHA may be due to (i) its short half-life following once-daily oral administration in mice (33), (ii) commencement of treatment at six weeks of age after the period of maximum linear growth velocity (34), and (iii) its potentially limited bioavailability in bone and cartilage. Overall, the current data suggest that SAHA is unlikely to have therapeutic benefit in the treatment of skeletal manifestations in RTHα.
In summary, the current studies demonstrate that TRα1 exerts major regulatory effects on linear growth and adult bone turnover that are independent of NCoR1. Overall, the findings suggest that the skeletal manifestations of RTHα are not mediated by persistent interactions between mutant TRα with NCoR1 and HDAC. The limited improvement of skeletal abnormalities in RTHα following disruption of the interaction between the dominant-negative TR and NCoR1 further suggests an important role for alternative co-repressors that interact with TR in skeletal cells such as silencing mediator of retinoid and thyroid hormone receptors (13).
Despite this, these studies identify a novel physiological role for NCoR1 in association with TRs to (i) optimize bone strength and (ii) limit excessive accumulation of cortical bone.
Footnotes
Acknowledgments
B.F. was supported by a Medical Research Council Clinical Research Training Fellowship (MR/P018718/1). A.N.H. received funding from a National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases R01 Grant (DK056123). S.-Y.C. is funded by NIH Intramural Research. J.H.D.B. and G.R.W. are funded by a Wellcome Trust Joint Investigator Award (110141/Z/15/Z and 110140/Z/15/Z).
Author Disclosure Statement
No competing financial interests exist for any of the authors.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4