
Abstract
Cell membrane isolation is essential for diverse biological investigations, ranging from fundamental research to advanced therapeutic applications. This study compared two methods—differential centrifugation and discontinuous sucrose density gradient ultracentrifugation—for isolating cell membranes from the human natural killer (NK) cell line (KHYG-1). The aim was to identify the method that minimizes contamination from nuclear, mitochondrial, and cytosolic components. Differential centrifugation yielded approximately 8 mg of cell membrane, whereas sucrose density gradient ultracentrifugation produced about 5 mg. Despite the lower yield, the latter method exhibited superior performance due to significantly reduced contamination. This protocol is adaptable to various cell types, offering a reliable approach for producing cell membrane-coated mimics for therapeutic use. The increasing demand for isolated cell membranes in biomedical applications highlights the importance of optimizing isolation techniques.
Impact Statement
This study evaluates two methods for isolating natural killer cell membranes, emphasizing the efficacy of sucrose density gradient ultracentrifugation in reducing contamination. The optimized technique can be applied to various cell types, supporting the development of cell membrane-coated mimics for therapeutic applications.
Introduction
The cell membrane, the outermost layer of a cell, is crucial for cell survival and proliferation, performing many complex functions.1,2 It comprises lipids, proteins, and carbohydrates. The lipid bilayer maintains the cell’s structural integrity, carbohydrates facilitate cell interfacing, and proteins are essential for signaling and adhesion.3–6 Specific protein markers on the cell surface are vital for various functions, including interactions and communication with other cells.
In recent years, the intrinsic functionality of cell membranes has been utilized in developing sophisticated delivery systems known as cell membrane-coated (CMC) mimics.7–9 These mimics capitalize on the unique properties of the cell membranes from which they are derived. Various types of cells, including nucleus-free10–14 and nucleus-containing cells,15–19 have been explored to design different CMC mimics.
Cell membrane isolation from leukocytes, specifically nucleus-containing cells, was initially discussed in 2013, utilizing discontinuous sucrose density gradient ultracentrifugation to design leukocyte membrane-coated mimics. 20 This method was later applied to various cell types for fabricating CMC mimics using either differential centrifugation17,21,22 or discontinuous sucrose density gradient ultracentrifugation.16,20,23 Despite the widespread use of these methods, a comprehensive comparison of their effectiveness in isolating pure cell membranes is lacking.
Nevertheless, the challenges associated with the isolation protocols of these membranes are often insufficiently addressed. Protocols for cell membrane isolation frequently lack specificity for different cell types, resulting in suboptimal outcomes.
Furthermore, numerous protocols fail to address the yield of membrane components or the preservation of functional integrity in isolated membrane proteins, often due to stringent isolation conditions. The scalability of these protocols is also a significant concern, limiting their application in large-scale preparations. Detailed procedural steps and critical parameters are frequently omitted, affecting reproducibility. Validation and verification steps to confirm purity and functionality are often insufficient, as are instructions for buffer preparation, handling, and storage. Comprehensive troubleshooting sections are typically lacking, and inadequate utilization of inhibitors during isolation can result in the degradation or modification of membrane proteins.
In this study, we focus on isolating cell membranes from KHYG-1 cells, an natural killer (NK) cell line, due to their surface resemblance to primary NK cells. These cells possess critical activating and adhesion receptors essential for targeting tumor and virally infected cells without prior activation. Moreover, KHYG-1 cells are easy to culture and expand in vitro, providing sufficient quantities of cell membranes for further use.24–28 Ensuring optimal growth conditions and maintaining cell health are critical in obtaining high-quality membranes for coating applications.
Utilizing the cell membranes from KHYG-1 cells to coat templates offers a promising approach for developing artificial NK cell mimicking systems to target tumor cells efficiently. 29 By leveraging the natural properties of these membranes, researchers can create advanced therapeutic delivery systems with enhanced targeting capabilities. It is crucial to isolate the membrane with minimal contamination to ensure that the maximum amount of receptor is translocated onto the template during the design of CMC mimics. This protocol emphasizes each step of the isolation process and provides troubleshooting tips to overcome common challenges. Through careful optimization and rigorous quality control, high-quality cell membranes can be obtained, paving the way for innovative biomedical research and therapy applications.
By refining the isolation process and understanding the underlying challenges, researchers can maximize the potential of CMC mimics, leading to more effective and targeted therapeutic strategies. Adjustment of the protocol with various hypotonic buffers and physical disruption techniques for different cell types has been recently published and reviewed. 30 This protocol is a valuable resource for scientists looking to harness the unique properties of cell membranes for various biomedical applications.
Materials and Methods
Preparation of KHYG-1 cell culture medium
To prepare the KHYG-1 cell culture medium, combine 500 mL of Roswell Park Memorial Institute1640 medium (RPMI 1640 with
Prepare heat-inactivated FBS by incubating 50 mL of FBS at 60°C for 30 min, followed by cooling, filtration, and storage at −20°C if not used immediately. Prepare human IL-2 stock solution (10 ng/µL) by dissolving 50 µg of IL-2 in 100 µL of sterile deionized water, vortexing, and aliquoting into 20 µL portions for storage at −80°C. Perform all IL-2 handling steps in a biosafety cabinet to maintain sterility.
Culturing and expansion of KHYG-1 cells
KHYG-1 cells (DSMZ, cat. no. ACC 725) were cultured and expanded over 2–3 weeks, starting with a vial of 4 × 106 cells in 8 mL of culture medium in a T25 flask with green ventilated cap (Sarstedt, cat. no. NC1272994) incubated vertically at 37°C. After 24 h, the cells were transferred to a T75 flask with green ventilated cap (Sarstedt, cat. no. NC9833320), remaining vertical for another day before being placed horizontally to reach 80% confluence. Cells were split into two T75 flasks with fresh medium containing IL-2, avoiding centrifugation unless the media appeared yellow, in which case cells were centrifuged at 28 g for 10 min before refreshing the medium. Upon subsequent splitting, cells were transferred to T175 flasks with green ventilated cap (Sarstedt, cat. no. NC1092669), maintaining density within 300,000–500,000 cells/mL. As cell growth accelerated, splitting was performed every 2 days, with density verified using a Bright-Line™ hemocytometer (Sigma-Aldrich, cat. no. Z359629). Once approximately 300 million cells were cultured, they were harvested by centrifuging at 112 g for 5 min in multiple 50 mL Falcon tubes. The collected pellets were pooled, washed with Dulbecco’s phosphate-buffered saline, and centrifuged at 112 g for 5 min, repeating the wash three times to remove residual medium. Around 16 T175 flasks were required to achieve the target cell count, with morphological changes observed in the KHYG-1 cells after centrifugation (Fig. 1A, B). After centrifugation of 300 million cells, cells tend to become more rounded due to cell clustering, increased cell-to-cell contact, limited space, and changes in the extracellular environment, all of which can impact cell signaling and cytoskeletal rearrangement.

KHYG-1 cells:
Preparation of hypotonic buffer for homogenization of cells
A hypotonic buffer is prepared by dissolving 23.80 mg of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Sigma-Aldrich, H3375), 31.31 mg of Potassium Chloride (KCl, Fischer Bioreagents, BP366), and 4.76 mg of Magnesium Chloride (MgCl2, Sigma-Aldrich, M8266) in 10 mL of deionized water. The pH is adjusted to 7.3 using 1N sodium hydroxide (NaOH). Prior to use, 20 μL of freshly prepared cOmplete™ ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor cocktail (Sigma-Aldrich, cat. no. 4693132001) is added per 2 mL of buffer to prevent the degradation of cell surface receptors.
The protease inhibitor cocktail is prepared by dissolving one tablet in 2 mL of deionized water, aliquoting the solution, and storing it at −20°C until required. The hypotonic buffer is always freshly prepared on the day of use and maintained on ice or at 4°C throughout the procedure.
Homogenization of the cells (summarized in Fig. 2)
Before homogenization, all materials, including the hypotonic buffer, Dounce homogenizer (tight pestle B, Sigma-Aldrich, cat. no. D8938), and 15 mL Falcon tubes, are prechilled in an ice bath for at least 15 min. The cell pellet is resuspended in 2 mL of ice-cold hypotonic buffer supplemented with 20 μL of protease inhibitor cocktail and incubated on ice for 30 min. The suspension is processed in two batches: 1 mL is transferred to the Dounce homogenizer, where 200 strokes are performed using the tight pestle B. The process is repeated with the remaining 1 mL. The homogenized mixtures are pooled into a single 15 mL Falcon tube, and a 10 μL sample is examined under a microscope to confirm cell lysis (Fig. 1C).
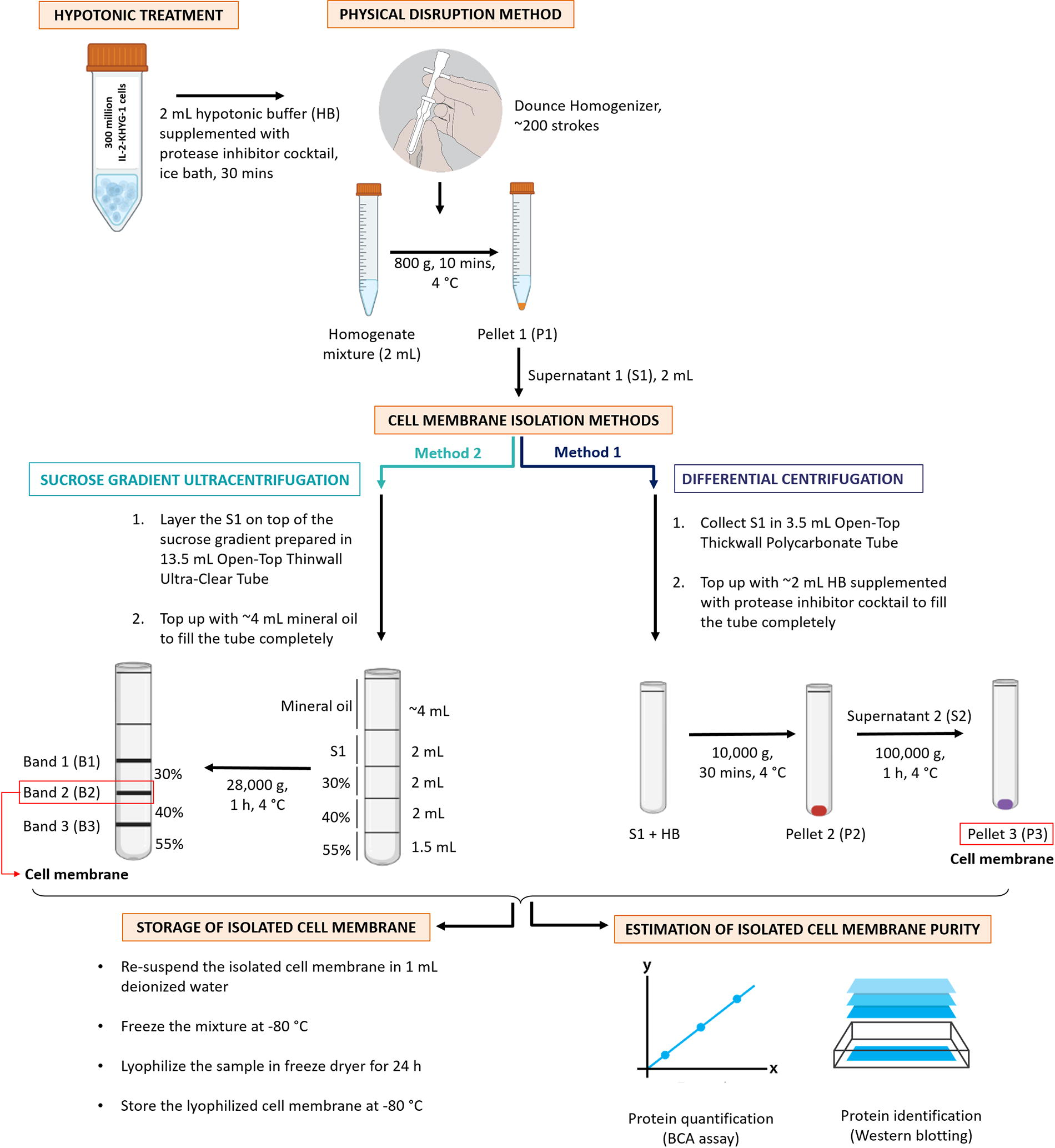
Schematic steps involved starting from the homogenization of cells till the isolation using both differential centrifugation and sucrose gradient ultracentrifugation; and storage of cell membrane followed with the estimation of its purity.
The homogenized sample is then centrifuged at 800 g for 10 min at 4°C in a precooled Multifuge X3R centrifuge. The resulting supernatant (∼2 mL, labeled S1) is collected for membrane isolation using differential centrifugation or sucrose density gradient ultracentrifugation. The centrifuge rotor is balanced with a tube of hypotonic buffer to ensure stability.
If microscopy reveals a significant number of intact cells, the pellet obtained after centrifugation at 800 g for 10 min is isolated, and homogenization is repeated on the pellet as needed. If homogenization is repeated, the total supernatant volume may increase to ∼4 mL. The remaining pellet is stored for subsequent analyses, such as western blotting.
Isolation of cell membrane
Cell membranes are isolated using either differential centrifugation or sucrose density gradient ultracentrifugation. Both methods involve prechilled equipment (SW55 Ti and SW41 Ti swinging-bucket rotor tubes) and the Optima™ XL-100 K ultracentrifuge set to 4°C. Rotor balancing and careful handling of the swinging bucket rotor are ensured throughout the process.
Method 1: Differential centrifugation
Two milliliters of the supernatant (S1) is placed into an open-top thick-walled polycarbonate tube (13 × 51 mm, Beckman Coulter, cat. no. 349622). The tube is filled with ∼2 mL of fresh hypotonic buffer containing a protease inhibitor cocktail to prevent tube stress that could cause failure. If S1 exceeds 2 mL (e.g., ∼4 mL), buffer addition is skipped, and centrifugation proceeds directly. The tube is placed into a prechilled SW55Ti rotor (Beckman Coulter, cat. no. 342194) and centrifuged at 10,000 g for 30 min at 4°C in the Optima XL-100 K ultracentrifuge (Beckman Coulter, cat. no. 8043-30-1208). The resulting supernatant (S2) is transferred to a new tube and centrifuged again at 100,000 g for 1 h at 4°C in Optima XL-100 K ultracentrifuge. The final supernatant is discarded, and the cell pellet, representing the isolated membrane, is retained for further analyses, such as western blotting.
Method 2: Discontinuous sucrose density gradient ultracentrifugation
To prepare the sucrose gradient, first make 0.9% (w/v) NaCl solution by dissolving 4.5 g of NaCl in 500 mL of deionized water, which can be stored at room temperature. Then, prepare sucrose solutions using this NaCl solution: dissolve 15 g of sucrose (Sigma-Aldrich, cat. no. S0389) in 50 mL for 30% (w/v), 20 g in 50 mL for 40% (w/v), and 27.5 g in 50 mL for 55% (w/v). Store these solutions at 4°C for up to 2 weeks. To assemble the gradient, use 2 mL syringes with long bevel needles (0.80 × 120 mm, B BRAUN) to layer the solutions sequentially into an open-top thin-walled ultra-clear centrifuge tube (14 × 89 mm, Beckman Coulter, cat. no. 344059): add 2 mL of 30% sucrose to the bottom of the tube without touching the walls, gently add 2 mL of 40% sucrose straight into the bottom of the 30% sucrose solution, and finally add 1.5 mL of 55% sucrose into the bottom of the 40% sucrose solution while maintaining distinct layers (as summarized in Fig. 3). Place 2 mL of supernatant (S1) on top of the 30% layer using a pipette. If S1 exceeds 2 mL (e.g., 4 mL), prepare two sucrose gradient tubes and distribute 2 mL of S1 in each, negating the need for a separate balancing tube. Add ∼4 mL of mineral oil to fill the tubes, ensuring structural integrity during centrifugation. Place the tubes in the precooled SW41Ti rotor (Beckman Coulter, cat. no. 333790) and centrifuge at 28,000 g for 1 h at 4°C. After centrifugation, collect the interface between the 30% and 40% sucrose layers using a syringe and needle. Transfer this interface into a polycarbonate tube, top with fresh hypotonic buffer containing a protease inhibitor cocktail, and balance the rotor with a hypotonic buffer-filled tube. Finally, centrifuge the collected interface at 100,000 using the SW55Ti rotor (Beckman Coulter, cat. no. 342194) for 1 h in Optima XL-100 K ultracentrifuge (Beckman Coulter, cat. no. 8043-30-1208). Discard the supernatant, and the resulting cell pellet is the purified cell membrane. Additional interfaces (e.g., sample—30% or 40–55%) may also be collected for other analyses, such as western blotting.
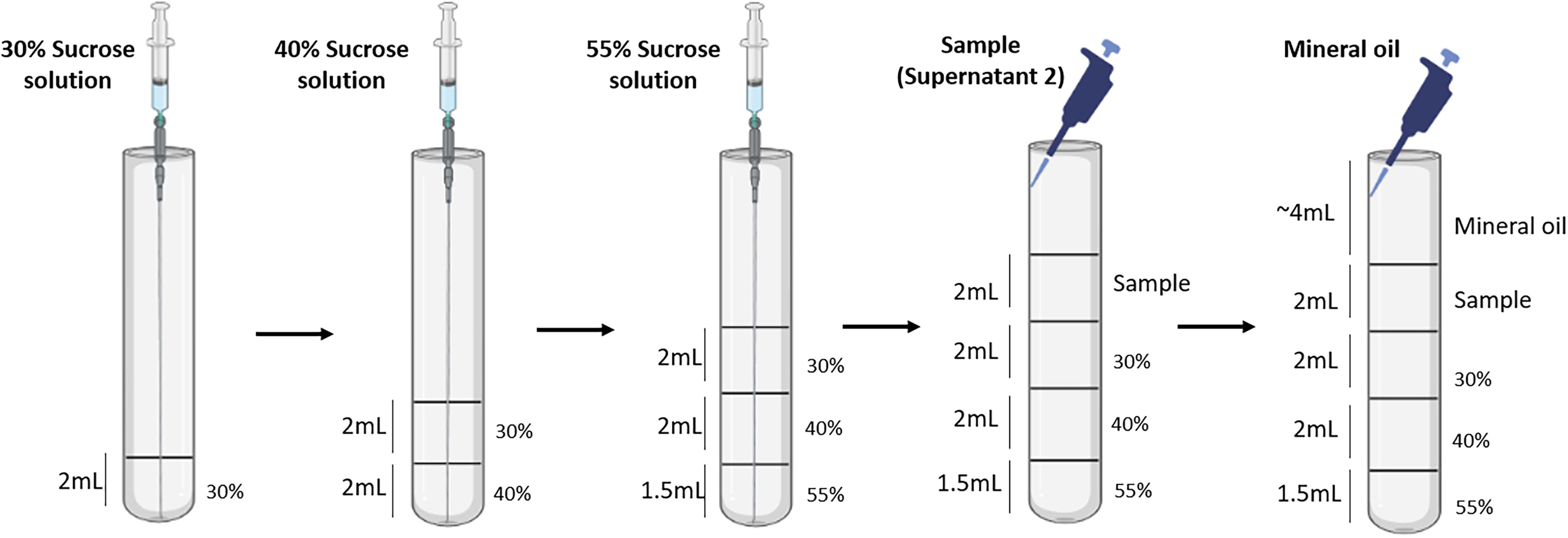
Schematic steps involved in preparing sucrose gradient using 2 mL syringe with long bevel needle and layering sample (S2) on the top with 1000 μL pipette.
Storage of isolated cell membrane
To store the isolated cell membrane, add 1 mL of deionized water to the cell membrane pellet and thoroughly disperse it using a pipette. Freeze the mixture overnight at −80°C, and then lyophilize it for 24 h using a freeze dryer. This process ensures the stability of cell surface proteins for extended use. The lyophilized membrane can be stored at −80°C for up to 6 months or at −20°C for 1 month if not used immediately. Each pellet obtained from differential centrifugation and bands collected during sucrose gradient ultracentrifugation can be similarly processed for further investigations, such as western blotting.
Experiment
The isolated KHYG-1 cell membrane was used to coat gelatin microspheres, creating NK cell mimics for targeting tumor cells, as described in our recently published article. 29 Sucrose gradient ultracentrifugation was employed for membrane isolation due to its minimal contamination, ensuring the maximum translocation of receptors onto the designed NK cell mimics.
Extraction and quantification of protein
Proteins were extracted from the isolated membrane by incubating 1 mg of each lyophilized sample (each pellet from method 1, each band from method 2) and 1 × 106 KHYG-1 cells in 70 μL of radioimmunoprecipitation assay buffer with protease inhibitor cocktail (1:100, cOmplete, Ethylenediaminetetraacetic (EDTA)-free, Roche, Inc., cat. no. 11873580001), phosphatase inhibitor cocktail (1:10, PhosSTOP™, Roche, Inc., cat. no. 04906845001), and phenylmethylsulfonyl fluoride (PMSF, 1:50, Sigma-Aldrich, cat. no. 93482) for 15 min on ice bath. Following incubation, the mixture was vortexed vigorously and then returned to ice for an additional 10–20 min.The mixture was subsequently centrifuged at 17,609 g for 15 min in picoTM 17 microcentrifuge (Thermo Fisher Scientific, cat. no. 75002401) at 4°C.The resulting supernatant was collected, aliquoted and stored at −80°C for further protein quantification and identification. The total amount of protein extracted from each sample was quantified by Pierce™ Bicinchoninic acid (BCA) Protein Assay Kit (Thermo Fischer Scientific, cat. no. 23225) to measure the absorbance at 562 nm. Different concentration of Bovine Serum Albumin (BSA, 0–1.2 mg/mL) is prepared from stock solution (2 mg/mL) for calibration curve.
Identification of proteins
The specific proteins were identified to determine the nuclear, mitochondrial, or cytosol contamination using western blotting. The antibodies used in this study include anti-nucleoporin p62 (BD Biosciences, cat. no. 610498), a mouse monoclonal antibody with a molecular weight of ∼62 kDa, employed as a nuclear marker. Anti-COX IV (Abcam, cat. no. ab14744), a mouse monoclonal antibody with a molecular weight of ∼17 kDa, serves as a mitochondrial marker. Anti-Glyceraldehyde 3-Phosphate Dehyrogenase (GAPDH, Abcam, cat. no. ab181603), a rabbit monoclonal antibody with a molecular weight of ∼36 kDa, is used as a cytosolic marker. Lastly, anti-CD11a (Abcam, cat. no. ab52895), a rabbit monoclonal antibody with a molecular weight of ∼180 kDa, is utilized as a KHYG-1 cell surface marker.
After the BCA assay, 5 µg protein from each sample (KHYG-1 cell lysate, each pellet from method 1, each band from method 2, isolated cell membrane from method 1 and method 2) was separated in a desired Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a Hybond® enhanced chemiluminescence (ECL)™ nitrocellulose membrane. Membranes were stained with Ponceau S to visualize successful transfer and protein loading. The membrane was blocked with 5% milk depending upon the primary antibodies’ suitability to avoid nonspecific binding. This was followed by specific primary antibody incubation overnight at 4°C and dilution as mentioned in Table 1. All washing steps were carried out in Tween-20 in tris-buffered saline (TBS, 0.1%). Next, horseradish peroxidase-conjugated secondary goat antirabbit or goat antimouse antibodies (prepared in 5% milk with dilutions mentioned in Table 1) were applied, followed by enhanced chemiluminescence detection. Signals from protein bands were captured on x-ray films (CLXPosure™ Film, Thermo Scientific™, cat. no. 34090).
Parameters for Using Primary and Corresponding Secondary Antibodies for Western Blotting Application
Results
Our protocol effectively isolates KHYG-1 cell membranes using a combination of a hypotonic buffer and a physical disruption technique (200 strokes of Dounce homogenizer), followed by differential centrifugation (M1) or sucrose density gradient ultracentrifugation (M2) methods. Protein was extracted from lyophilized pellets (P1, P2, P3) obtained from different centrifugation steps (800 g, 10,000 g, and 100,000 g) in differential centrifugation and distinct bands (B1, B2, B3) in sucrose gradient ultracentrifugation using the same lysis buffer for further purity estimation. Based on protein quantification using the BCA assay (Fig. 4), it revealed concentrations of approximately 1.961 ± 0.016 mg/mL for P1, 2.77 ± 0.004 mg/mL for P2, and 2.970 ± 0.030 mg/mL for P3, 1.761 ± 0.047 mg/mL for B1, 1.643 ± 0.019 mg/mL for B2, and 1.905 ± 0.066 mg/mL for B3. Furthermore, purity and cell surface protein analysis in each pellet and band were performed using semiquantitative analysis (western blotting) to confirm the most suitable regions for isolated cell membranes (Fig. 5). According to the protein identification for nuclear content, mitochondrial content, and cell surface marker, P3 was deemed optimal for differential centrifugation, containing no mitochondrial content, some nuclear content, and a substantial CD11a receptor presence. Conversely, B2 was identified as the ideal region for sucrose gradient ultracentrifugation due to its absence of mitochondrial content, limited nuclear content, and increased CD11a receptor concentration. Notably, the typical yield of lyophilized cell membrane was approximately 8 mg for P3 from M1 and 5 mg for B2 from M2.
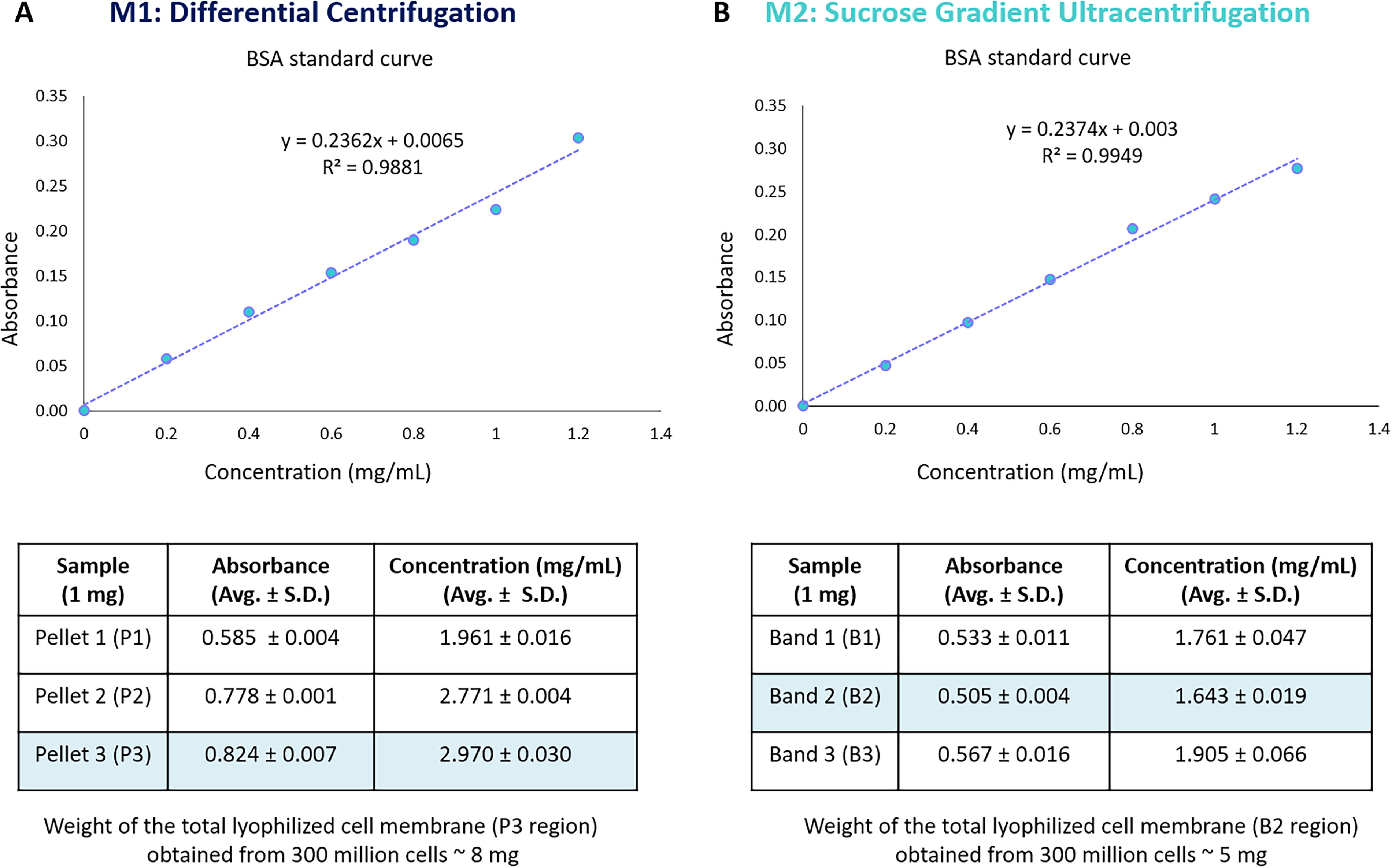
Total protein quantified in each 1 mg lyophilized pellet P1, P2, and P3 [in case of differential centrifugation
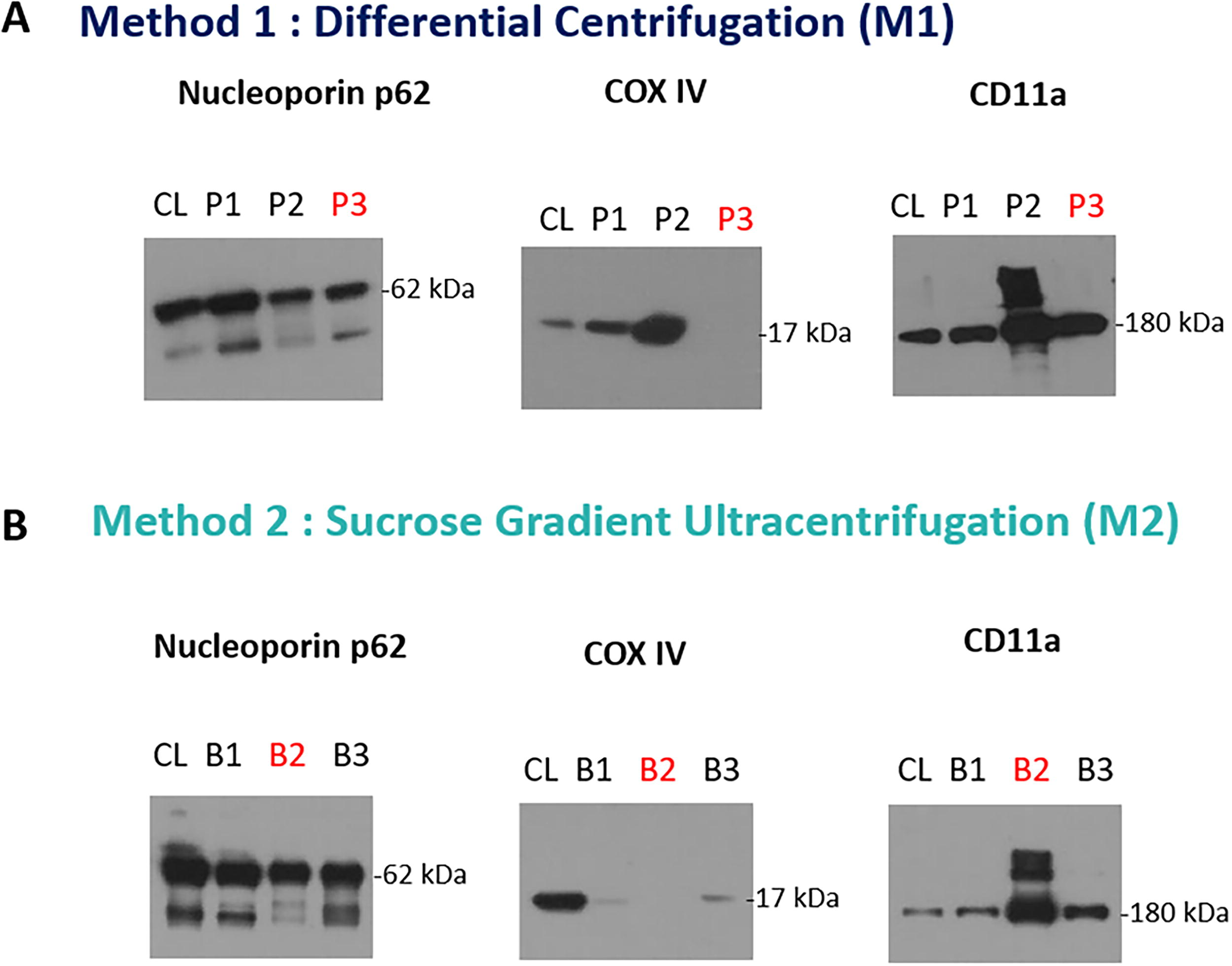
Western blot analysis to estimate specific protein marker in each pellet [in case of differential centrifugation method
Although the protein content and yield for P3 were approximately double those of B2, it was insufficient to conclude. Therefore, a comparative analysis of both methods was conducted to determine the approach with the most negligible contamination. As illustrated in Figure 6, M2 exhibited reduced nuclear and cytosolic content alongside more CD11a receptor presence compared with M1. Notably, both M1 and M2 showed no mitochondrial contamination. Consequently, M2 (sucrose gradient ultracentrifugation) was identified as the superior method for isolating cell membranes with minimal contamination.
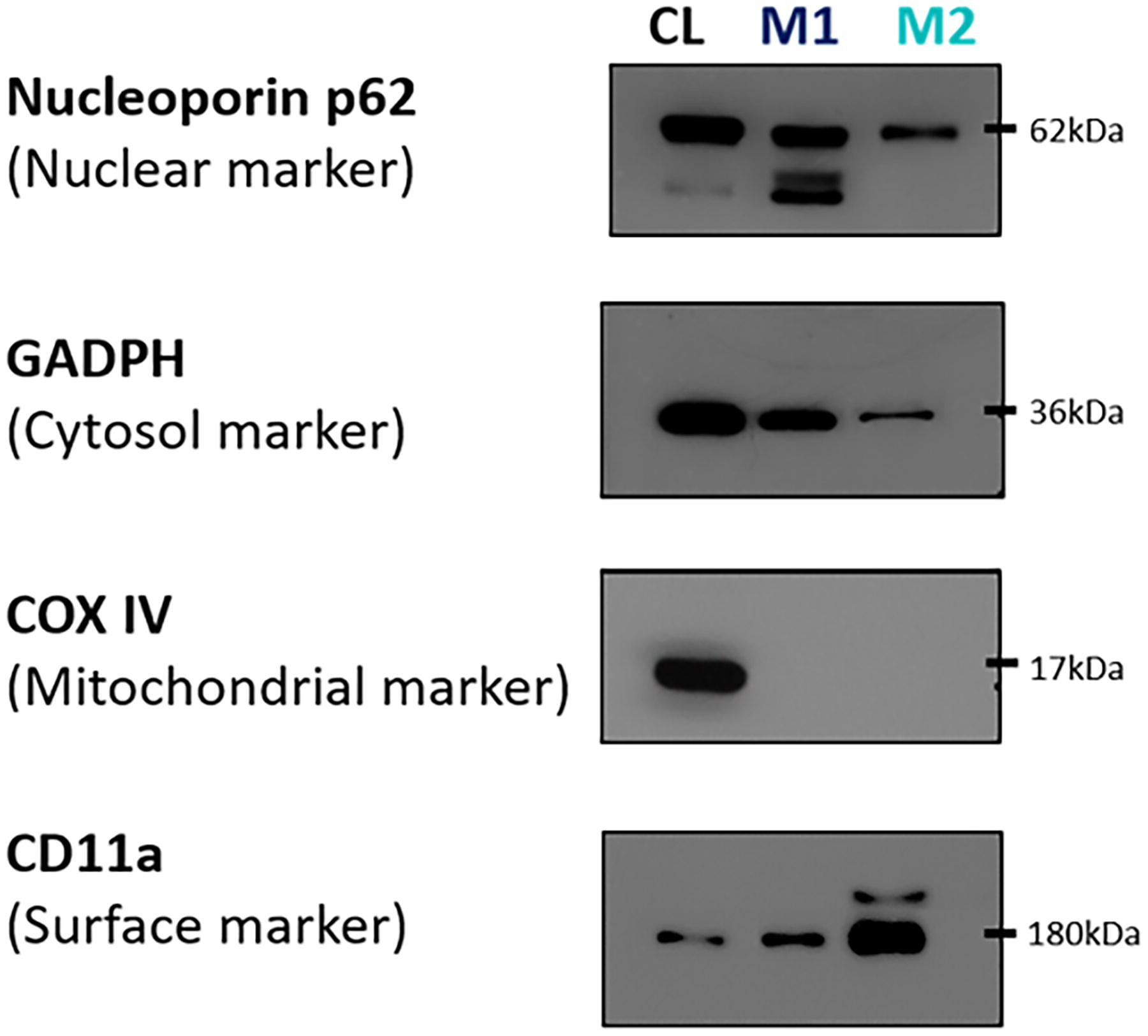
Comparative analysis of the purity of the isolated membrane obtained from both methods: differential centrifugation (M1) and sucrose gradient ultracentrifugation (M2) using western blotting. M2 has less nuclear cytosol content and more cell surface receptors than M1. Both M1 and M2 have no mitochondrial contamination. Therefore, M2 is the best method to isolate cell membranes with minimal contamination.
This protocol can also be adapted for isolating membranes from other nuclear-containing cells with appropriate modifications. The selection of hypotonic buffers and physical disruption techniques, as reported in our recent review, provides a foundation for tailoring protocols to specific cell types if needed. 30 To ensure reproducibility, each step of the isolation process has been detailed, with troubleshooting strategies outlined in Table 2 to address common challenges.
Troubleshooting Tips to Overcome Common Challenges During Cell Membrane Isolation
The table outlines common challenges encountered during cell membrane isolation, along with their potential causes and suggested solutions. These strategies aim to improve the reproducibility and yield of the isolated cell membrane.
Discussion
Cell membrane isolation is a crucial step in developing CMC mimics and other biomedical applications such as drug delivery, vaccine development, and immunotherapy.16,31,32 Two commonly used techniques for isolating cell membranes are differential centrifugation and sucrose gradient ultracentrifugation, each with distinct advantages and limitations. Despite the widespread use of differential centrifugation and sucrose gradient ultracentrifugation for membrane isolation, their comparative effectiveness remains insufficiently explored.16,17,20,21,33–36 This study aimed to evaluate these two techniques in terms of membrane yield, purity, and practical feasibility using KHYG-1 NK cells as a model system. Selecting the appropriate method is essential to ensure optimal membrane yield, purity, and functionality for downstream applications.
Differential centrifugation is one of the most commonly used methods for cell membrane isolation due to its simplicity, accessibility, and ability to process large volumes of cells efficiently. The method relies on multiple centrifugation steps at increasing speeds to sequentially separate cellular components based on size and density. This stepwise process enables the enrichment of cell membranes while removing larger organelles, such as nuclei and mitochondria, through lower-speed spins. The primary advantage of differential centrifugation lies in its high yield, making it a practical choice when a large amount of membrane material is required, such as in large-scale studies, industrial applications, or high-throughput screening. Additionally, the equipment needed for this method—standard centrifuges with variable speed settings—is widely available in most laboratories, eliminating the need for specialized ultracentrifugation devices. The shorter processing time compared with sucrose gradient ultracentrifugation also makes it an attractive option when rapid isolation is necessary. However, despite its efficiency in membrane recovery, differential centrifugation suffers from significant limitations regarding membrane purity. Due to its reliance on size and density differences alone, this method often results in contamination from nuclear, mitochondrial, and cytoplasmic components, which can compromise the functionality of the isolated membranes. This contamination poses a particular challenge for applications that require high specificity, such as receptor-ligand interaction studies or targeted drug delivery systems, where even minor impurities can interfere with experimental outcomes. Additionally, the repeated high-speed centrifugation steps may induce mechanical stress, potentially leading to membrane disruption, protein denaturation, and loss of functional receptors. The presence of residual cellular debris and organelles in the final membrane fraction can also lead to inconsistencies in experimental reproducibility, making optimization and validation critical when using this method.
Sucrose gradient ultracentrifugation offers a more refined approach to membrane isolation by utilizing a discontinuous density gradient to separate cellular components based on their buoyant densities. This method is particularly valuable in applications where membrane purity is paramount, such as in functional studies of membrane-bound receptors, immunotherapy development, and precision drug delivery. Unlike differential centrifugation, which relies solely on differences in sedimentation rates, sucrose gradient ultracentrifugation provides a more selective separation by allowing membranes to migrate to specific density layers, effectively reducing contamination from other organelles. The major advantage of this technique lies in its superior ability to yield highly pure membrane fractions with minimal contamination from nuclear and cytoplasmic components, ensuring that functional membrane proteins remain intact. This purity is critical when working with sensitive downstream applications that require precise biological activity, such as biochemical assays, proteomics, and targeted nanoparticle coating for drug delivery. Additionally, the density-based separation mechanism results in more consistent and reproducible membrane isolation, which is beneficial for comparative studies and large-scale research efforts.
However, despite its advantages, sucrose gradient ultracentrifugation has notable drawbacks. The technique is significantly more time-consuming and labor-intensive compared with differential centrifugation, as it requires careful preparation of gradient layers, prolonged ultracentrifugation steps, and meticulous fraction collection to avoid contamination between layers. The requirement for an ultracentrifuge, which is both costly and not universally available in all research settings, also limits the accessibility of this method, particularly for laboratories with limited resources. Additionally, while the purity of isolated membranes is enhanced, the overall membrane yield is lower than that obtained through differential centrifugation. This lower yield can be a significant drawback when working with cells that are difficult to culture in large quantities or when a substantial amount of membrane material is needed for subsequent applications. Another challenge of sucrose gradient ultracentrifugation is the technical expertise required to perform the separation correctly; improper gradient preparation or inaccurate fraction collection can lead to inconsistent results, affecting reproducibility and experimental success. Moreover, prolonged ultracentrifugation at high speeds may cause structural alterations in certain membrane proteins, potentially impacting their functionality, although this risk is lower compared with the mechanical stress imposed by differential centrifugation. Despite these challenges, the ability of sucrose gradient ultracentrifugation to yield highly pure and functionally intact membranes makes it an indispensable method for studies where membrane composition and integrity are critical.
Despite the effectiveness of both techniques, there remains a need for further optimization to improve the scalability, reproducibility, and consistency of the methods. The complexity of membrane isolation, combined with the variability in cell health and preparation conditions, poses challenges in ensuring high-quality membrane samples for all types of downstream applications. The introduction of automated systems, improved buffer compositions, and advanced centrifugation equipment may help overcome some of these limitations, offering the potential for more efficient, high-throughput isolation of cell membranes.
After the isolation of cell membranes, proper storage is crucial to maintaining their stability, functionality, and protein integrity. While freezing is a widely used preservation method, improper handling can lead to structural damage and functional loss. Suspending them in phosphate-buffered saline (PBS) with protease inhibitors helps prevent protein degradation and enzymatic activity loss. Repeated freeze-thaw cycles can cause membrane rupture, protein denaturation, and loss of receptor functionality, compromising their intended applications. Additionally, slow freezing may result in ice crystal formation, which could disrupt the membrane stability. To mitigate these effects, rapid freezing in liquid nitrogen (−196°C) or dry ice is recommended to prevent large ice crystal formation. For short-term storage (days to weeks), membranes should be stored at −20°C in small aliquots to avoid repeated freeze-thaw cycles, which accelerate degradation. For long-term storage (months to years), membranes should be kept at −80°C, preferably after rapid freezing in liquid nitrogen or dry ice to preserve structural integrity. One of the best long-term preservation methods without freezing is lyophilization (freeze-drying), which removes water from the membrane to prevent crystal-induced damage, allowing for storage at ambient temperatures. However, rehydration is a critical step in restoring membrane integrity, and it must be performed carefully to avoid aggregation or protein denaturation. This can be achieved through slow pipetting, low-speed vortexing, or mild sonication in an appropriate cold buffer (such as PBS) on ice to prevent heat-induced protein denaturation. Additionally, allowing membranes to equilibrate at 4°C for atleast 30 mins before use in sensitive applications ensures proper recovery of functional properties. Freeze-drying is an excellent method for long-term storage and transport of cell membranes, especially for drug delivery systems, biosensors, and vaccine development. However, it is not suitable for applications requiring native membrane fluidity like electrophysiology and ion channel studies, membrane fusion and vesicle studies, and live-cell integration. Regardless of the storage method, western blotting or SDS-PAGE should be used to assess membrane protein stability and integrity poststorage. By implementing optimized freezing techniques, and careful rehydration methods, researchers can effectively preserve membrane quality for future applications while minimizing degradation.
Conclusion
Both differential centrifugation and sucrose gradient ultracentrifugation offer valuable approaches to cell membrane isolation, with their effectiveness depending on the specific requirements of the study. Differential centrifugation is best suited for applications that require high membrane yield and rapid processing, although it comes with the drawback of contamination from other cellular components. It is an ideal method when large-scale membrane isolation is needed and when absolute purity is not the primary concern. On the contrary, sucrose gradient ultracentrifugation provides superior purity and minimizes contamination, making it the method of choice for applications that require functionally intact membrane proteins and high specificity. However, its technical complexity, longer processing time, and lower yield present challenges that must be carefully managed. The selection between these methods should be guided by the balance between purity and yield required for the intended application. Moving forward, further refinements in isolation protocols, such as incorporating automated gradient formation systems or developing hybrid techniques that combine the strengths of both methods, may enhance the efficiency and scalability of cell membrane isolation, paving the way for more advanced biomedical and therapeutic applications.
Footnotes
Acknowledgment
The authors would like to acknowledge Mr. Anthony Sloan’s editorial assistance.
Authors’ Contributions
V.C.: Authored the article, wrote the article, troubleshoot, and executed all the experiments and optimizations. V.K.K.: Technical support in troubleshooting, organizing, and analysis of the data. V.P.: Technical support for western blotting. A.P.:Supervision, project administration, funding acquisition, conceptualization, editing.
Data and Code Availability
The protocol includes all datasets generated or analyzed during this study. The study did not develop new unique codes.
Disclosure Statement
The authors declare no competing financial interests.
Funding Information
This publication has emanated from research conducted partly by a grant from Science Foundation Ireland (SFI) and the European Regional Development Fund (ERDF) under grant number 13/RC/2073_P2. V.K.K. has received funding from SFI Industry Fellowship under grant number 19/IFA/7447. V.P. has received funding from SFI and the ERDF under grant number 13/RC/2073.