
Abstract
Stem cell products derived from mesenchymal stem cells (MSCs) have been widely used in clinical trials, and a few products have been already commercialized. However, the therapeutic effects of clinical-grade MSCs are still controversial owing to mixed results from recent clinical trials. A potential solution to overcome this hurdle may be to use clonal stem cells as the starting cell material to increase the homogeneity of the final stem cell products. We have previously developed an alternative isolation and culture protocol for establishing a population of clonal MSCs (cMSCs) from single colony forming unit (CFU)-derived colonies. In this study, we established a good manufacturing practice (GMP)-compatible procedure for the clinical-grade production of human bone marrow-derived cMSCs based on the subfractionation culturing method. We optimized the culture procedures to expand and obtain a clonal population of final MSC products from single CFU-derived colonies in a GMP facility. The characterization results of the final cMSC products met our preset criteria. Animal toxicity tests were performed in a good laboratory practice facility, and showed no toxicity or tumor formation
Introduction
S
Materials and Methods
Isolation and culture of cMSCs
Bone marrow aspirates were obtained from the iliac crest of three healthy donors after written informed consent (approved by Inha University Hospital Institutional Review Board; IRB number 10–51). Isolation of cMSCs was performed as described previously. 9 In brief, small human bone marrow aspirate was mixed with 15 mL of isolation medium: Dulbecco modified Eagle Medium containing low glucose (Gibco-BRL, Life Technologies, Gaithersburg, MD), and with 20% fetal bovine serum (FBS; Gibco-BRL) and 1% penicillin/streptomycin (Gibco-BRL), and then incubated in a 100-mm culture dish. As shown in Figure 1A, after incubation for 2 h at 37°C with 5% CO2, only the cell culture supernatant was transferred to a new 100-mm dish. After the second 2-h incubation, the supernatant was again transferred to a new dish (D1) and incubated for additional 2 h. The supernatant was transferred to another new dish (D2) and incubated for a day, and then transferred to another new dish (D3) and incubated for a day. This process was repeated two more times with 1- and 2-day incubations (D4 and D5, respectively). In the first three transfers after a short period of incubation, we wanted to let high-density cells, such as white and red blood cells, settle to the bottom of the dish, and the progressively longer intervals between the supernatant transfers would allow the less dense and/or adhesive cells in the supernatant to settle to the bottom of the dish. We hypothesized that this method might allow cells with different densities and/or adherences to be further fractionated. Only well-separated single colonies with a diameter of about 4 mm, ranging from ∼4 × 103 to 1 × 104 cells in a single colony, in D2, D3, D4, or D5 dishes were selected and transferred to six-well plates and then to larger culture flasks, where they kept expanding. After 10–14 days in the 100-mm dishes, the single colonies were detached and isolated using a 1- to 2-min treatment with 0.05% trypsin/EDTA (Gibco-BRL) treatment in cloning cylinders (). Once the cells reached 70–80% confluence, they were recovered with trypsin/EDTA and replated for further expansion. For cryopreservation, cells harvested were thoroughly washed, filled with cryopreservation medium, which is composed of 10% pharmaceutical-grade dimethyl sulfoxide (OriGen Biomedical, Korea) and 90% FBS, and then cryopreserved in a liquid nitrogen tank until thawing.
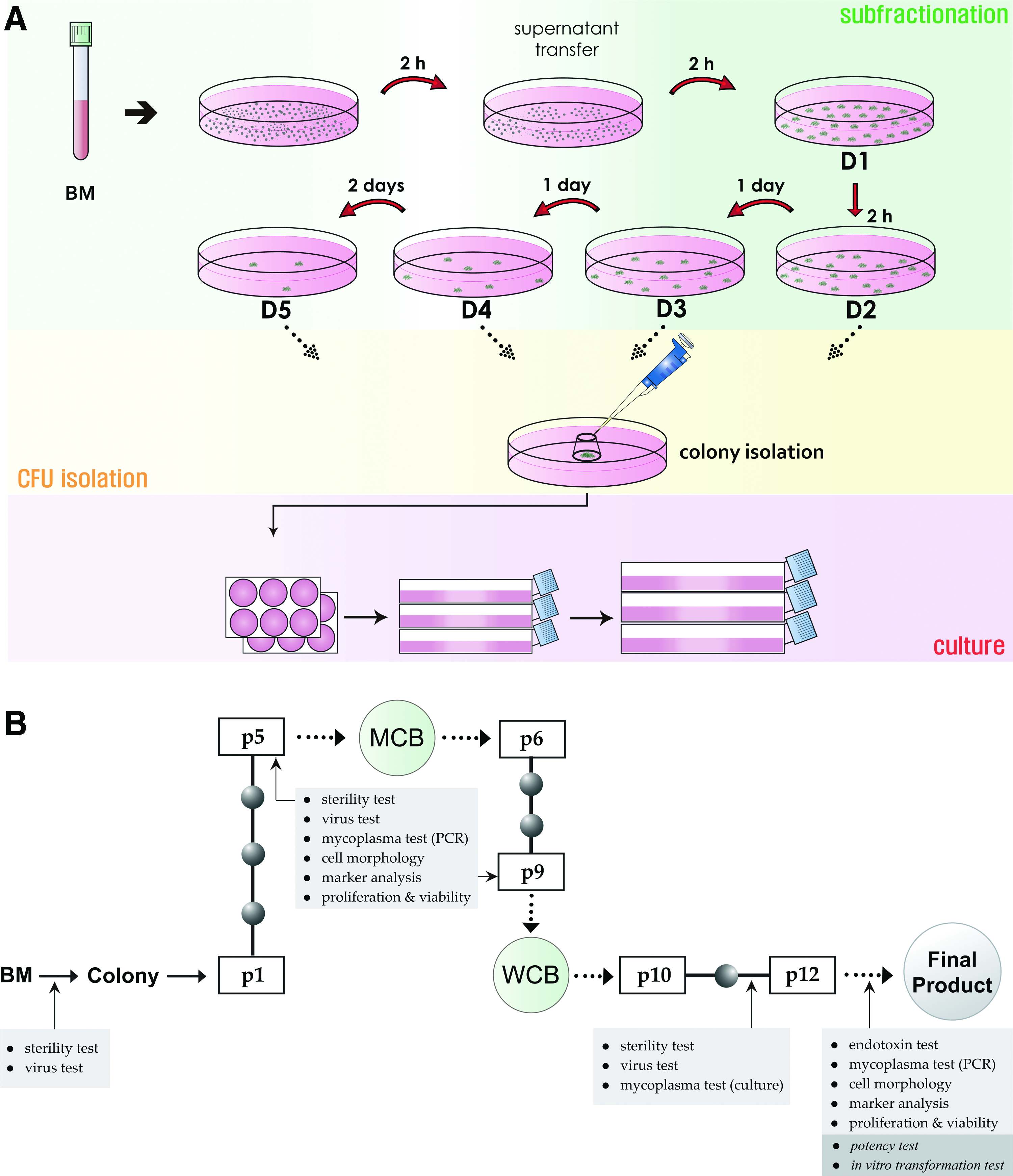
Schematic representation of clinical-grade cMSC production and quality control processes in a good manufacturing practice facility.
Due to space limitations, the other parts of materials and methods including cMSC characterization, toxicity tests, and sterility tests are described in Supplementary Materials and Methods (Supplementary Data are available online at
Results
Establishment of a GMP-compatible SOP for the manufacture and quality control of cMSC production based on SCM
SCM is capable of producing MSC clones from small aspirates of bone marrow as reported previously.9,17 The key issue was whether our laboratory-scale SCM was applicable to GMP-compatible manufacturing procedures to produce sufficient number of cell products for patients in a reproducible manner. We optimized the laboratory-scale SCM protocol to be suitable for large-scale expansion in a GMP facility (Fig. 1A). For MSC isolation from bone marrow aspirates, the basic process was not different from the original protocol. Briefly, the bone marrow aspirates from the ileac crest of a healthy donor were mixed gently and thoroughly in the isolation medium. Repeated transfers of the supernatants containing floating bone marrow cells after the heavier cells settled down (taking up to 5 days) were performed in a 100-mm culture dish. Colonies with high CFU activities were selectively isolated by trypsinization in a cloning cylinder, and they were allowed to grow in a six-well plate. This step was determined as passage 1. After the growing cells reached about 70% confluence, the cells were detached and plated to grow in a 75-cm2 culture flask (passage 2). From this step, the culture time for a passage was set within 72 to 120 h. Further cell expansion was done in a 175-cm2 culture flask at a seeding density of 0.5–1 × 106 cells/175-cm2. Features of this GMP-compatible manufacturing process were two-point banking steps. Master cell bank (MCB) was the first cell freezing point for ensuring potential cell clones with good proliferation activity. Working cell bank (WCB) was planned for the second cell freezing point, from which the final cell products were to be produced. Cells obtained at passages 5 and 9 were cryopreserved in liquid nitrogen for MCB and WCB, respectively. The final products were produced from cells harvested at passage 12. Because GMP should ensure the quality and safety of the final cell products for the patients, a strictly-regulated quality control process was established in addition to the manufacturing process (Fig. 1B). When screening the source material, the presence of pathological microbes and viruses was checked (Refer to Supplementary Materials and Methods for details). At passage 5 before the MCB storage, tests for pathologic microbes and mycoplasmas were performed, during which the fast assay for cytopathic effect (CPE) and hemadsorption were performed instead of the virus test. CPE, which refers to the morphological change in the host cells caused by a viral infection, was assayed by culturing cMSC-treated human normal lung fibroblast cell line MRC-5 and monkey kidney epithelial cell line, Vero, for up to 14 days to determine the infection. Measles virus and sendai virus were used as control viruses. The hemadsorption assay checked the ability of the host cells, infected by the hemagglutinin-producing virus, to adsorb erythrocytes. At the same time, the cultured cMSCs were checked for the homogeneous fibroblast-like cell morphology, MSC marker analysis for MSC identification and purity, and cell viability. The cells for MCB storage must meet all of the preset criteria. If the cells fail to meet any of preset specifications, the cells are discarded immediately. At passage 9 before WCB storage, the same tests as for MCB storage were performed. Normal virus test was added at this step. A third test was done between passages 11 and 12. As for the source material screening, sterility and virus tests were performed. During this process, mycoplasmas were additionally screened by the culture method. When the final cell products were released from passage 12, the existence of endotoxin and mycoplasma were checked. In parallel, the quality of the cultured cells was tested in terms of cell morphology, MSC marker analysis, and cell viability.
Characterization of clinical-grade cMSC products
Two clones named cMSC1 and cMSC2 were used for characterization. Our standards for MSC identification and characterization included four criteria: cell morphology, cell surface marker expression, differentiation potential, and immunosuppression. Plastic-adherent cMSCs exhibited fibroblast-like shapes. They retained fibroblastic morphology at passages 9 and 12 (Fig. 2A), indicating that the morphological consistency of cMSCs is maintained during all the passages of subculture. Second, the cell surface marker expression was analyzed using flow cytometry. Two clones were positive for CD29, CD44, CD73, CD90, CD105, CD146, CD166, and HLA-Class I, but were negative for CD14, CD31, CD34, CD45, CD106, CD119, CD184, and HLA-DR (Table 1). Each cell surface marker showed a constant expression at different passages (9 and 12) and between different clones, indicating a homogeneous population of cells during the production process.
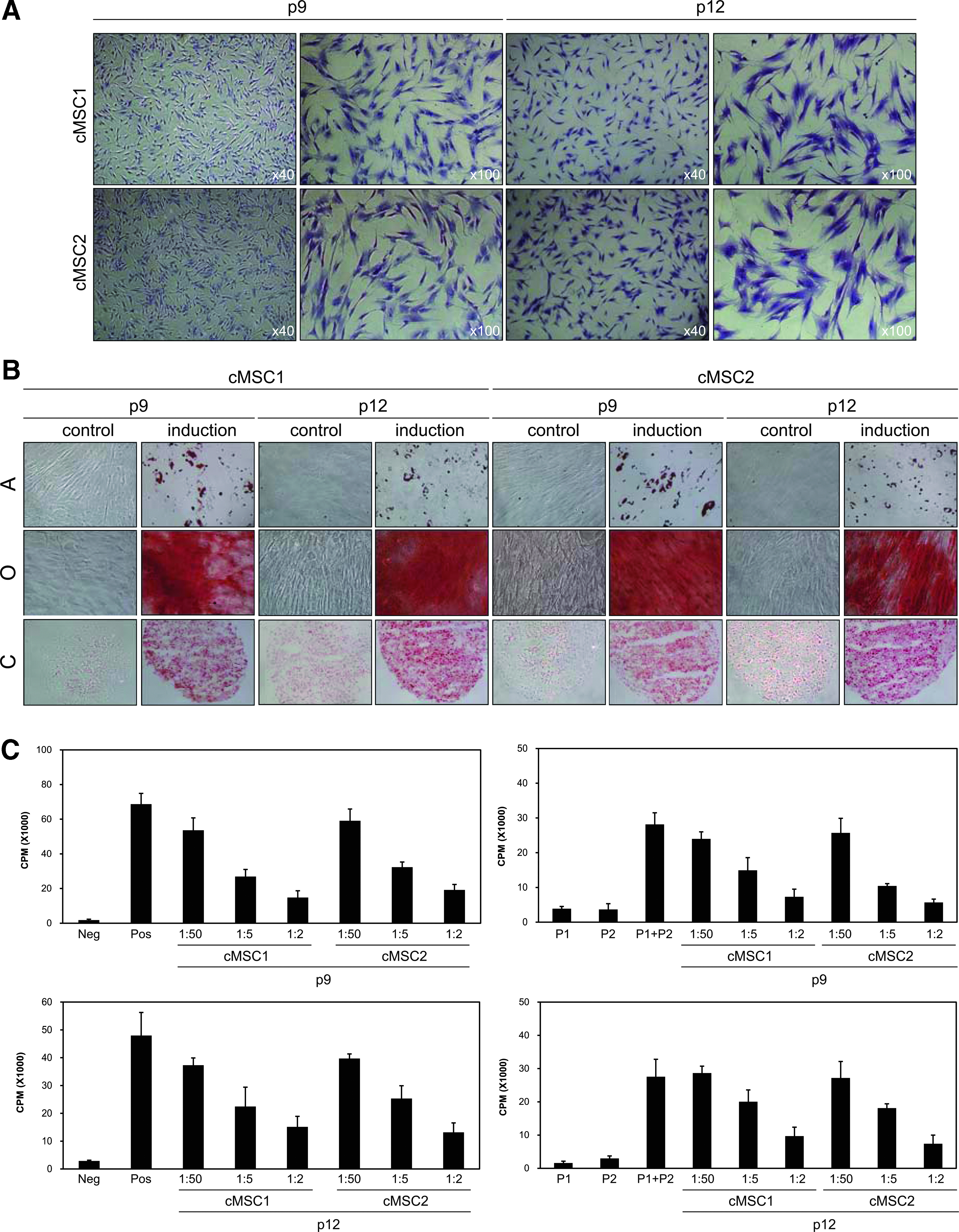
Characterization and properties of clinical-grade cMSCs.
cMSC, clonal mesenchymal stem cell.
Next, the differentiation potential of these cMSCs was examined. Although over-cultured MSCs reportedly show reduced differentiation ability, we harvested cells at late passages (9 and 12) to induce them to differentiate into the three mesenchymal cell types. The induced cells at both passages were determined by tissue-specific staining (adipocytes visualized using oil red O, osteoblasts by alizarin red S, and chondroblasts by safranin O) (Fig. 2B). The results showed unchanged differentiation capability of each clone even at passage 12, which was further supported by molecular marker expression for each differentiation (Supplementary Fig. S1). Finally, the
Safety evaluation of clinical-grade cMSC products
One of the main objectives of GMP production is to provide safe products for patients. For the safety of the final products, we evaluated their toxicity in immunodeficient athymic nude mice (Table 2). The toxicity test was performed in a certified good laboratory practice facility. The tests comprised single injection toxicity, multiple injection toxicity, biodistribution and cell detection, and tumorigenicity
NOAEL, no observed adverse effect level.
A single injection toxicity test was designed to evaluate the acute toxicity of TS. Three doses of cMSCs (5 × 104 cells/300 μL normal saline for injection/head for low-dose, 5 × 105 for medium-dose, and 1 × 106 for high-dose) were injected into the female and male mice, and then the mice were observed up to 14 days. During the observation period, there were no deaths in the control and stem cell-treated groups. Neither clinical abnormality nor significant weight change was evident. Necropsy of all animals at 14 days showed no gross findings, indicating no acute toxicity of the cMSC products (Table 2).
The multiple injection toxicity test was conducted with the low, medium, or high doses of TS (Table 2). Each dose of TS was injected three times at an interval of 2 weeks, based on the required time for production of large amounts of the final cMSC products from WCB cells. For acute toxicity evaluation, all mice were examined for 4 weeks for clinical signs including body weight, food consumption, ophthalmological examination, urinalysis, hematological examination, clinical chemistry, necropsy for gross pathology, organ weights, and histopathology. No death was observed. There were no effects on body weights, food consumption, and ocular abnormalities during the observation period. No change in urinalysis, hematology, and clinical chemistry data associated with the cMSC injection was observed throughout the course of the test. No cMSC-related effects on organ weights were evident in any animals. Furthermore, no gross finding or lesion in necropsy and histopathological examination was considered attributable to TS. In mitogen-induced lymphocyte proliferation assays, splenic T and B lymphocytes remained unchanged in males and females in the treated groups as compared with the control group. For long-term toxicity evaluation, we examined other sets of control and high-dose groups with a recovery period of 6 weeks later, following third administration of the high-dose TS. During the recovery period, one male in the control group died on day 63 and no histopathological finding could clearly indicate the cause of death. No abnormal clinical sign was found in TS-treated group. There was no cMSC-related change in body weight, food consumption, ocular abnormality, hematology, and clinical chemistry. In necropsy, a black focus in the glandular stomach was observed in one male of the control group, which was caused by mucosal necrosis according to the histopathological analysis. A white focus in the heart was observed in another male in the same group. Small testes and a white focus in the heart were in one male of the cMSC-treated group. According to the histopathological examination, the small testes were associated with atrophy of seminiferous tubules, while the white focus in the heart was epicardial mineralization. However, there was no toxicological significance because of the incidental and sporadic occurrence. Local irritancy at the injection sites was not evident in any animal in TS-treated group. In mitogen-induced lymphocyte proliferation assays, proliferation of splenic T and B lymphocytes was not altered in any animals of the recovery groups treated with TS compared with control group. Collectively, there was no cMSC-related effect on mice at the high dose. The results indicated that the observed adverse effect level (NOAEL) of the cMSC products is smaller than 1 × 106 cells/head in mice under the conditions of this test (Table 2).
To trace the
Tumorigenicity is considered to be one of the most important hurdles in the clinical application of stem cells.18,19
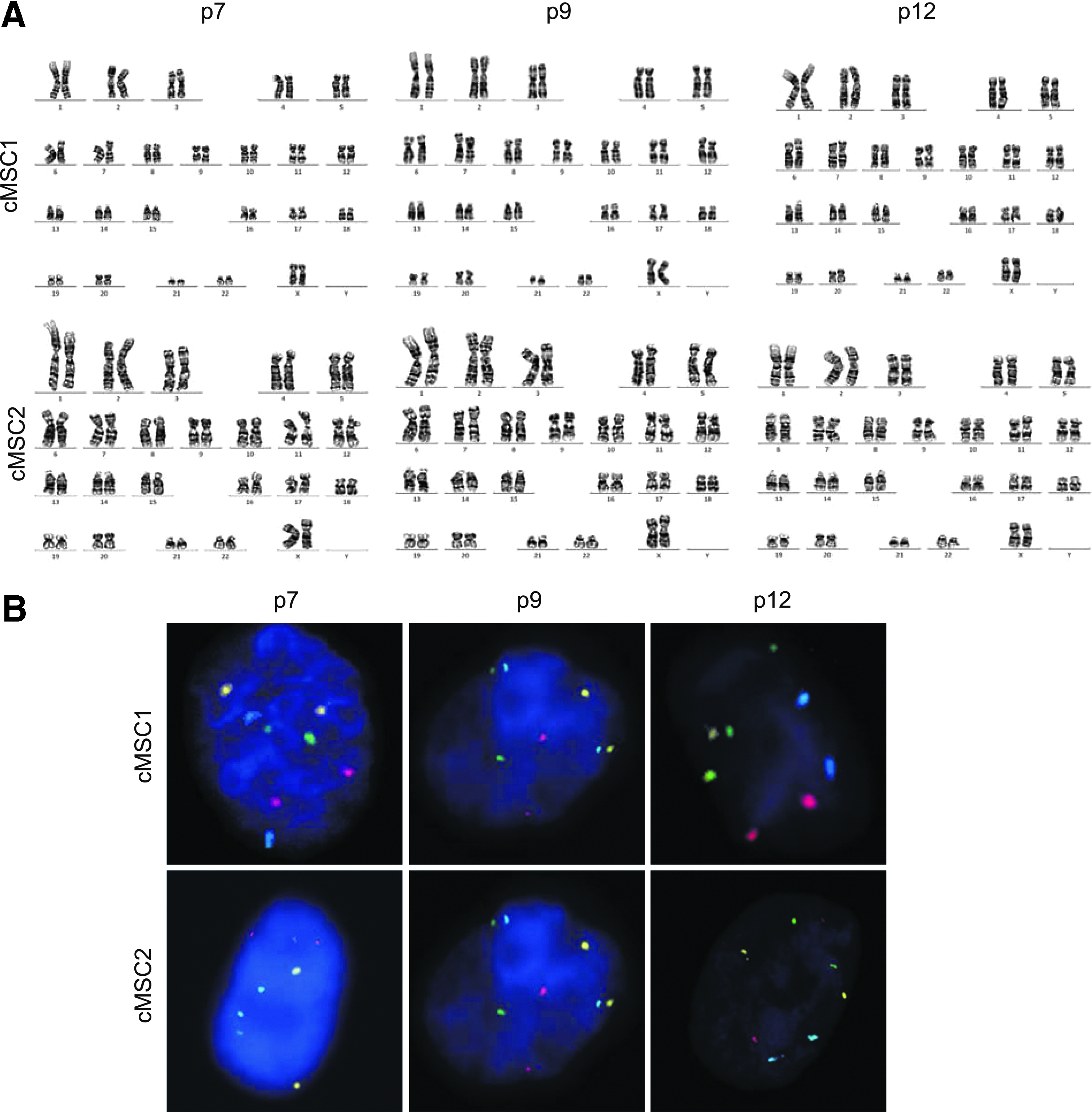
Chromosomal analysis of clinical-grade cMSCs.
Practical production of clinical-grade cMSCs
We next sought to determine the amount of cMSC products that can be practically manufactured using our GMP-compatible methodology based on the SCM. Three independent trials were conducted. As shown in Table 3, there was substantial variation in colonies that were isolated for further expansion among bone marrow donors. Among these colonies, 20–30% of the clones that fully met the MCB criteria were deemed to be suitable for MCB cryopreservation. Furthermore, only 25–45% of these MCB-suitable clones fulfilled the WCB requirements. The other clones that did not fulfill the requirements for the MCB or WCB were dropped out of the manufacturing process. However, the stock numbers of cell vials at the WCB were not proportional to the number of WCB clones, because the proliferation activity of each clone differed. For example, production batch-1 produced only 4 WCB clones, which in turn produced 1430 WCB cell vials. On the other hand, batch-2 produced 10 WCB clones, but only 719 cell vials were ultimately cryopreserved for the WCB due to the relatively lower proliferation activity of this batch compared with that of batch-1. This suggests that upon release of these cells to patients as the final products, a total of 474 product packages (5 × 107 cells per package per 50 kg human; the prospected clinical dose of cMSCs is 1 × 106 cells/kg body weight of a patient) could be manufactured from three batches of cMSC production. Thus, about 470 patients (1 product per 50 kg patient) could be treated with this batch production. These data indicated that our GMP methodology is sufficient to produce cMSC products from small aspirates of the bone marrow for treating a large number of patients.
BM; MCB, master cell bank; WCB, working cell bank.
Discussion
In this study, we established a GMP-compatible manufacturing procedure for the clinical-grade production of human bone marrow-derived cMSCs based on the SCM (Fig. 1). We optimized the culture procedures to expand and obtain a population of cMSCs from single CFU-derived colonies. The characterization results of the cMSCs in the MCB, WCB, and final products met our preset criteria of morphology, cell surface antigen expression, differentiation potential, and suppression of lymphocyte proliferation (Fig. 2 and Table 1). The results of the animal preclinical toxicity tests showed no toxicity and no tumor formation
There are still several main hurdles to overcome in stem cell therapeutics with MSCs. These include the improvement of efficacy, setting up a standard manufacturing protocol, reducing the cost of large production, and elucidation of the treatment mechanisms. The clinical trials performed over the last decade have showed the safety of clinical-grade MSCs but mixed clinical outcomes have showed both optimistic and pessimistic prospects about the efficacy of MSC products.4–6,21,22 One of the possible reasons for mixed results may be the heterogeneity of the final MSC products. Due to the lack of MSC-specific isolation of the current isolation methods, the final MSC products produced by the conventional methods may contain heterogeneous populations of cells. A potential solution to overcome this hurdle may be to use cMSCs as the starting cell material to maximize the homogeneity of the final stem cell products. Bianco
It is unfeasible to obtain a homogeneous population of MSC products with current isolation and culture technologies. Production of homogeneous MSC products is not theoretically possible even when individual clones of MSCs are used as the starting material, mainly because stem cells divide asymmetrically during the long mass culture process, and all differentiation- and senescence-related events are kinetically asymmetric. To date, no attempt has been made to produce MSC products with single CFU-derived colonies, probably because it is assumed that not enough cells can be produced to meet the demands of clinical trials. Here, we proved that a sufficient number of cMSC products can be manufactured with single CFU-derived colonies. This mass production of cMSCs using SCM was an unexpected result. A very large number of cMSCs can be produced by this standard manufacturing system in the absence of any special culture procedures or devices. Because a low percentage (5–10%) of cell colonies showed such expansion capability, it is possible that the “true” MSCs have a much higher proliferation capacity than that of normal primary cells. In the future, we aim to compare the proliferative potential of cMSC lines
Toxicological evaluation of stem cell products is very important step to determine the degree of toxicity and the relationship between the dose and adverse effects of stem cells administered. This evaluation also provides information on target organs and target functions, allowing for a scientifically supported extrapolation of the potential effects of these products in humans. In particular, the characterized risks need to be extrapolated to clinical situations and patients. The quality and reproducibility of safety data are key components of the utility of stem cell products to support the assumption of safety in humans. Our
As a technical note, the procedure applied here to generate cell colonies does not account for the density of cell plating at each supernatant transfer. It is possible that not all colonies were clonal populations originating from single cells. In particular, during the final passages of supernatant transfer, the floating cells could be dividing cells from the previous dish rather than small and/or less adherent cells. Therefore, an improved design would help set a precise density of cells at each supernatant transfer rather than just transferring the equally divided volume of supernatant; this would ensure a more systemic and effective selection of the clonal origin of each line.
The advantages of using a population of cMSCs for stem cell therapy and tissue regeneration are the following: First, it could reduce the possibility of causing immune reactions because other cell types are not contaminated. 26 Second, it may increase the efficacy of MSC products, similar to more pure chemical drugs that have higher treatment efficacy. Third, patients may need a smaller number of MSCs to treat diseases because a population of cMSCs is used. Fourth, eliminating centrifugation, enzymatic treatment, and filtering procedures to obtain MSCs could lower the cost for large production. Fifth, a library of cMSC lines can be established and a specific MSC line having a greater potential to treat specific diseases or to induce tissue regeneration can be chosen. Overall, cMSC products could be a more efficacious and economic option in clinical settings.
Therapeutic stem cells are living drugs and are different from chemical drugs. The fundamental difference is that the components of the chemical drugs can be controlled whereas there is a limit to the regulation of stem cells. Due to this kind of inevitable limitation, it has been generous to developers of stem cell therapeutics to allow the use of heterogeneous stem cells. One solution for manufacturing highly “homogeneous” stem cell products is to use a population of clonal stem cells at the beginning of the manufacturing process. Additional efforts to manufacture more homogeneous stem cell products for their better safety and efficacy should be pursued. Here, we suggest a potential alternative to achieve this goal.
Conclusions
In this study, we established a GMP-compatible procedure for the clinical-grade production of human bone marrow-derived cMSCs based on the SCM. We optimized the culture procedures to expand and obtain a clonal population of final MSC products from single CFU-derived colonies in a GMP facility. The characterization results of the final cMSC products met our preset criteria. Animal toxicity tests showed no toxicity or tumor formation
Footnotes
Acknowledgments
This study was supported by the Bio &Medical Technology Development Program (NRF-2011-0019634 & NRF-2011-0019637) of the National Research Foundation by the Korean government (MEST), and by a grant from Inha University (44773-01).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.