
Abstract
Endothelial cells (ECs) are widely used in research, both for fundamental vascular biology research and for exploring strategies to create engineered vascularized tissues. Primary isolation often results in contamination from fibroblasts and vascular smooth muscle cells that can potentially affect function, particularly during the initial expansion period needed to establish the cell culture. In the current study, we explored the use of microcarriers to selectively isolate ECs from the lumen of intact vessels to enhance the purity during the isolation procedure. First, rat aortic explant culture was performed and after 2 weeks of culture, flow cytometry revealed that only 60% of the expanded cell population was positive for the endothelial marker CD31. Then, we employed a strategy to selectively isolate ECs and improve their purity by introducing microcarriers to the lumen of intact aorta. After 10 days, microcarriers were carefully removed and placed in cell culture dishes and at 15 days, a large near confluent layer of primary ECs populated the dish. Flow cytometry revealed that >90% of the expanded cells expressed CD31. Moreover, the cells were capable of forming tubule-like structures when plated onto Matrigel, confirming their function also. The highly modular and transportable nature of microcarriers has significant potential for isolating ECs at high purity, with minimal contamination.
Introduction
V
ECs have been isolated from a range of different species and from anatomically heterogeneous locations, and cell types commonly employed for studies include umbilical vein ECs, umbilical artery ECs, aortic ECs, and microvascular ECs (often from the dermis due to ease of accessibility) 4 as well as endothelial progenitor cells,5,6 all of which have properties that can be exploited for vascular biology research.7,8
Several methods can be adopted to isolate ECs, including enzymatic digestion, combined mincing and enzymatic digestion, and mechanical scraping or physical detachment of ECs from the underlying basement membrane. 4 Enzymatic digestion using collagenase detaches the EC sheet from the underlying basement membrane and the resulting cell population displays high purity. It is the classical method used for isolating human umbilical vein ECs, 9 but a major disadvantage is that proteolytic digestion can potentially damage key transmembrane proteins, as can the centrifugation and trituration required to derive a single cell suspension. Combined mincing and digestion brings inherent contamination problems as mincing vascular tissue will result in a mixed population of cells with potential contaminants being vascular smooth muscle cells, fibroblasts, and pericytes, depending on the vessel origin. Mechanical scraping of ECs from large vessels, such as the aorta, 10 or physical detachment in microvasculature by perfusion with microcarriers in a cold buffer11,12 can yield different endothelial populations that are high purity but low yield. Furthermore, the extent of damage caused by the physical abrasion or cold shock, respectively, is not fully characterized. Finally, explant cultures can be performed that are particularly useful for arterial EC isolation.
Artery walls consist of three layers: tunica adventitia, tunica media, and tunica intima. ECs reside in the innermost layer of the tunica intima where they are in direct contact with the blood. In explant culture, blood vessel tissue is dissected and placed endothelial side down, so that ECs in contact with the culture dish can migrate out of the explant. However, poor dissection techniques may introduce contaminating fibroblasts or vascular smooth muscle cells from the outer layers of the vessel. Moreover, fibroblasts are highly motile and are capable of migrating from the upper layer as part of the outgrowth population, further affecting the purity of the EC population. 13
Microcarriers that support cell attachment and growth have enormous potential for achieving scalable cell expansion that is required to meet the clinical demand for cell therapies.14–16 In addition, given the highly modular and transportable nature of these substrates for cell growth, microcarriers might enable a suitable alternative methodology to be adopted for the high-purity isolation of ECs. Indeed, early studies utilized microcarriers to isolate the microvascular endothelium using a perfusion method with cold saline and ethylenediaminetetraacetic acid (EDTA) to release ECs from the basement membrane onto the microcarrier surfaces. 11
In the current study, we demonstrate that primary isolation of rat aortic outgrowth ECs using the explant culture method is suboptimal for isolating ECs due to the high level of contamination present. We then demonstrate that a newly established method that entails introducing poly(ɛ-caprolactone) (PCL) microcarriers to the lumen of the intact aorta can isolate populations of ECs at significantly improved purity.
Materials and Methods
Animals
This study was approved by the Animal Care and Use Committee of Dankook University. Four male Sprague Dawley rats were used.
Materials
Polymer and solvents used for the microsphere preparation
PCL (Mw=80,000; Sigma-Aldrich), chloroform (Sigma-Aldrich), poly(vinyl alcohol) (PVA; Sigma-Aldrich), and 70% ethanol.
Reagents used for the primary culture
Hanks' balanced salt solution (HBSS; Gibco), penicillin (100 U/mL)/streptomycin (100 mg/mL; Sigma-Aldrich), Vascular Cell Basal Medium (ATCC, cat # PCS-100-030), Endothelial Cell Growth Kit (ATCC, cat # PCS-100-041), Dulbecco's phosphate-buffered saline (DPBS; Gibco), 0.25% trypsin/1 mM EDTA (Gibco-BRL), dimethyl sulfoxide (DMSO; Sigma-Aldrich), and Matrigel™ (BD Bioscience).
Tools used for dissecting vascular and cell seeding
Large scissors, dissecting scissors, large forceps, dissecting forceps, 200 μL yellow pipette tips (ø: 1049.23±1.39 μm/540.1±0.97 μm), 1000 μL blue pipette tips (ø: 731.23±1.19 μm/399.88±1.2 μm), and an insulin syringe.
Cell culture vessels
Thirty-five-millimeter dishes, 60-mm dishes, and 15-mL falcon tubes.
Imaging devices
Dissecting microscope, Samsung digital camera, inverted microscope (Olympus), ZEISS M700 confocal laser-scanning microscope (CLSM), and scanning electron microscope (SEM; Hitachi S-3000H).
Fabrication of PCL microspheres
The PCL was dissolved in chloroform within a vial at 10 wt.% while stirring for 3 h. The mixture solution was dropped within a water pool containing 1 wt.% PVA while stirring at 600 rpm at 4°C. The ratio of the mixture solution to the water bath was determined as 1:30. The mixture solution became solid rapidly on dropping into the ice-cooled bath, and after the solidification, the solution was further stirred for an additional 6 h to harden the microspheres. The hardened microspheres were washed with ice-cooled distilled water five times through filtration with CHM® Ashless Filter papers (weight: 85 g/m2, thickness: 180 μm). The obtained PCL microspheres were further freeze dried and kept at 4°C for further characterization and use.
Cell culture procedure
Segments of thoracic aortae (22–25 mm) were excised from four male Sprague Dawley rats (190–250 g) and immediately placed in cold HBSS. This solution was utilized throughout the entire tissue dissociation procedure. Clotted blood and connective tissue were dissected from the outer wall of the vessels under the 10× magnification dissection microscope. Any remaining blood residues were removed by flushing the lumen of the vessels with a small volume of HBSS. Then, the vessel was immersed in the Vascular Cell Basal Medium supplemented with the Endothelial Cell Growth Kit.
Fifteen milligrams of microspheres was sterilized with 70% ethanol for 2 h and washed with the PBS solution three times. The microspheres were prewetted in the culture medium for 12 h. The prewetted microspheres were placed in the vessel lumen and cultured for 10 days. Then, the microspheres were moved into a 35-mm dish and cultured for further 10 days. On day 11, the cells were trypsinized and transferred into 60-mm culture dishes and cultured continuously for 5 days.
Cell morphology observation and proliferation assay
To confirm the attachment and the distribution of the cells on the microspheres, the specimens were stained using Alexa Fluor 546-conjugated Phalloidin. Briefly, the specimens were rinsed with 1 mL of 1× PBS and fixed with 4% paraformaldehyde for 15 min at room temperature (RT), then permeabilized in 0.2% Triton X-100 in 1× PBS for 5 min, and blocked with 1% (w/v) BSA in 1× PBS for 30 min to prevent nonspecific protein binding. The specimens were incubated with 20 nM Alexa Fluor 546-conjugated Phalloidin diluted in 1× PBS for 30 min at RT, washed with 1 mL of 1× PBS three times for 5 min each at RT. The specimens were covered with a mounting medium. A fluorescence image was obtained using an Olympus IX71 inverted microscope equipped with a DP-72 digital camera.
To quantify the proliferation of the cells, cell counting was performed using the trypan blue exclusion test. Briefly, a uniform suspension containing the cells was added to the trypan blue dye by a 1:1 dilution. The number of viable cells was obtained by counting the cells of four 1×1 mm2 squares of a hemocytometer and averaging. The total cell number of viable (unstained) cells was calculated directly as mean values of viable cells. Each experiment was repeated three times.
Characteristic analysis of the primary EC culture
The characteristics of the ECs were confirmed by tube formation and immunofluorescence staining for the von Willebrand factor (vWF). For tube formation assay, 50 μL of thawed Matrigel solution was coated onto each well of a four-well sterile plate and incubated for 30 min at 37°C to allow the Matrigel solution to form a gel. HUVECs were seeded at a density of 2×104 cells onto Matrigel and grown in the Vascular Cell Basal Medium with the EC Growth Kit for 1 day.
For vWF immunofluorescence staining, 3×104 cells were directly seeded in 12-well plates with coverglass. After 3 days, the medium was removed and the cells were fixed by incubating each well with 1 mL of 4% paraformaldehyde for 10 min at RT. The PFA was aspirated and the wells were washed once with 1 mL of 1× PBS at RT. The 1× PBS was discarded, the cells were incubated for 30 min at RT in a blocking buffer (1% bovine serum albumin in 1× PBS), and permeabilization was achieved by incubation with 0.1% Triton X-100 for 5 min. The blocking buffer was removed and the primary antibody vWF solution (1:200 dilution in 1× PBS, cat # ab6322; Abcam) was added. The mixture was incubated in the dark for 60 min at RT. The primary antibody solution was aspirated and the wells were washed with 1 mL of 1× PBS three times for 5 min each at RT. The secondary antibody goat anti-rabbit Alexa Fluor 555 solution (1:150 dilution in 1× PBS, cat # A21428; Invitrogen) was added and incubated for 30 min at RT in the dark. The cells were washed with 1 mL of 1× PBS three times for 5 min each at RT. For nuclei counterstaining, the wells were incubated with 0.5 mg/mL 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) for 5 min at RT. The DAPI was removed and the coverglass containing the cells was mounted on the slide with two drops of mounting medium. The borders of each slide were covered with nail polish to fix the coverglass to the slide. A fluorescence image was taken on the ZEISS M700 CLSM.
Flow cytometry analysis
The cells were suspended in 1 mL 1× PBS at ∼1×106 cells/mL. To detect EC-surface antigens, a 100-μL of the cell suspension was put into a flow cytometry tube, a primary antibody against PECAM-1 (CD31; cat# sc-80913; Santa Cruz Biotechnology, Inc.) was added into the tube at 1 μg/mg and incubated for 30 min at 4°C. The cells were washed with 2 mL of 1× PBS and pelleted by centrifugation at 1500 rpm for 5 min at RT. The supernatant was discarded and the cell pellet was resuspended with 100 μL of 1× PBS. The fluorescein isothiocynate-conjugated secondary antibody (cat# sc2010; Santa Cruz Biotechnology, Inc.) at 1 μg/mL was added into the tube and incubated for 30 min at 4°C in the dark. The cells were washed with 2 mL of 1× PBS and pelleted by centrifugation at 1500 rpm for 5 min at RT. The supernatant was discarded and the cell pellet was resuspended with 0.5 mL of 1× PBS. Labeled cells were analyzed by a flow cytometer (FACS Calibur; Becton Dickinson) and Cell Quest software (Becton Dickinson).
Statistical analysis
Data are shown as the mean±one standard deviation. Statistical comparisons were made using the Student's t-test. p<0.01 was considered to be statistically significant.
Results
Purity of expanded isolations of outgrowth ECs from rat aortic tissue explants
To confirm that contamination from non-EC types from the arterial wall (Fig. 1A, B) results from explant culture, segments of rat aorta were placed lumen side down in cell culture dishes and incubated for 9 days to enable cell outgrowth. Cells were then cultured for further 14 days to obtain an established culture of cells that could then be assessed by flow cytometry. Flow cytometry using an antibody to CD31 (PECAM) revealed that only 60% of cells within the population were positive for the EC marker (Fig. 1C).
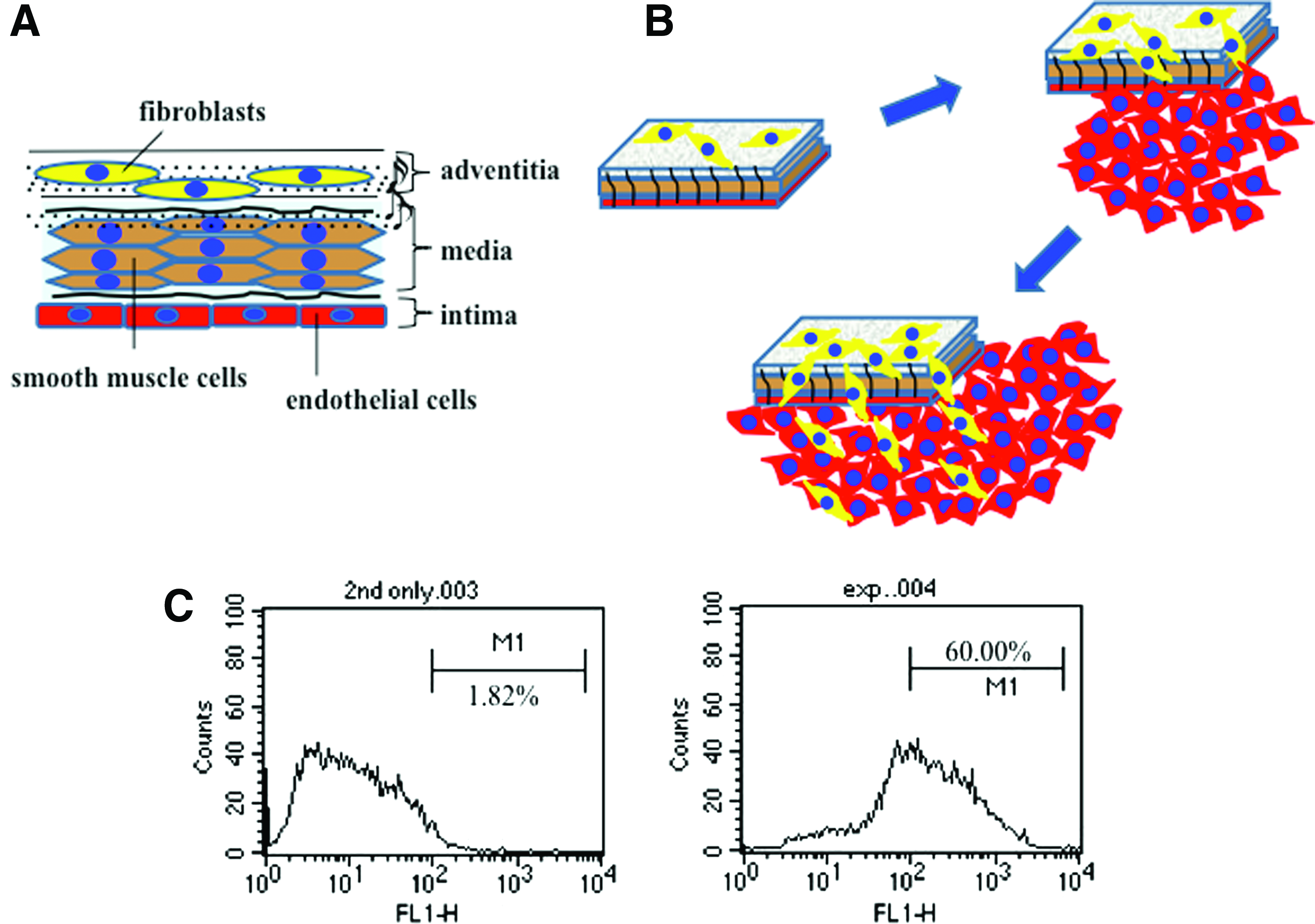
Contamination in cultures of outgrowth endothelial cells (ECs) from vessel explant tissue.
Microcarrier scaffold characterization
The PCL microcarriers produced as described previously 17 characterized using SEM to demonstrate their morphology (Fig. 2A). They had a relatively broad size distribution (Fig. 2B) and those within the range 200–500 μm (average diameter=386 μm) were used for cell culture.

Poly(ɛ-caprolactone) (PCL) microcarrier characteristics.
Microcarrier isolation of cells
Standard dissection tools were used to prepare thoracic aortas (Fig. 3A) and the aortas typically had a length of around 22–25 mm (Fig. 3B). After loading the aortas with microcarriers, they were maintained as organ cultures ex vivo, (as illustrated in Fig. 3C). Retrieval of cells on microcarriers was accomplished using a yellow pipette tip (Fig. 3D). The basic concept of utilizing microcarriers placed in the lumen of an intact aorta as a strategy to improve EC purity at isolation is presented in Figure 4. It was expected that if microcarriers were introduced to the vessel lumen through a pipette and tissue culture performed, cells would migrate onto the microcarrier substrates and populate them over time.
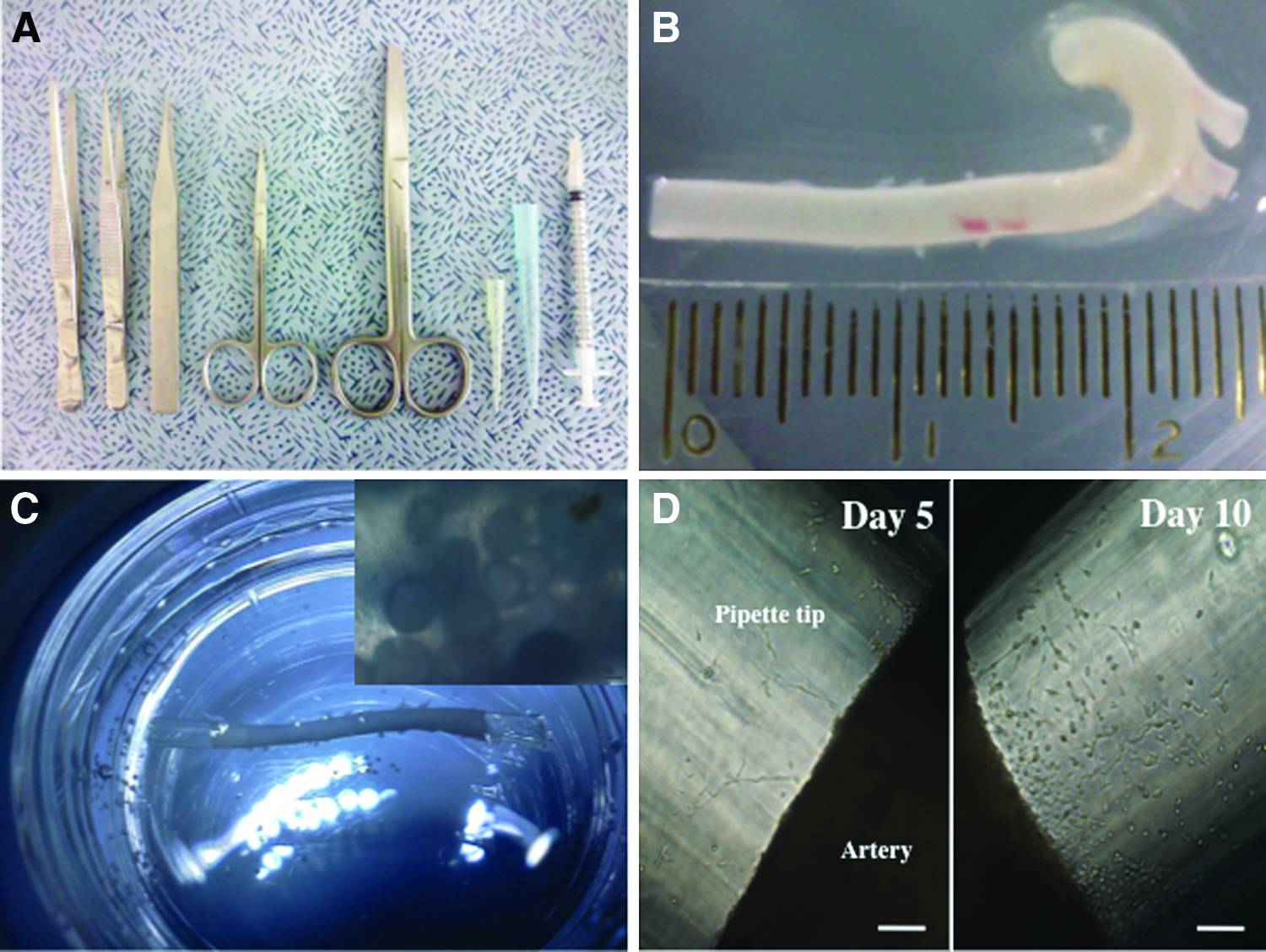
Equipment for the preparation of microcarrier-loaded aortic organ cultures.
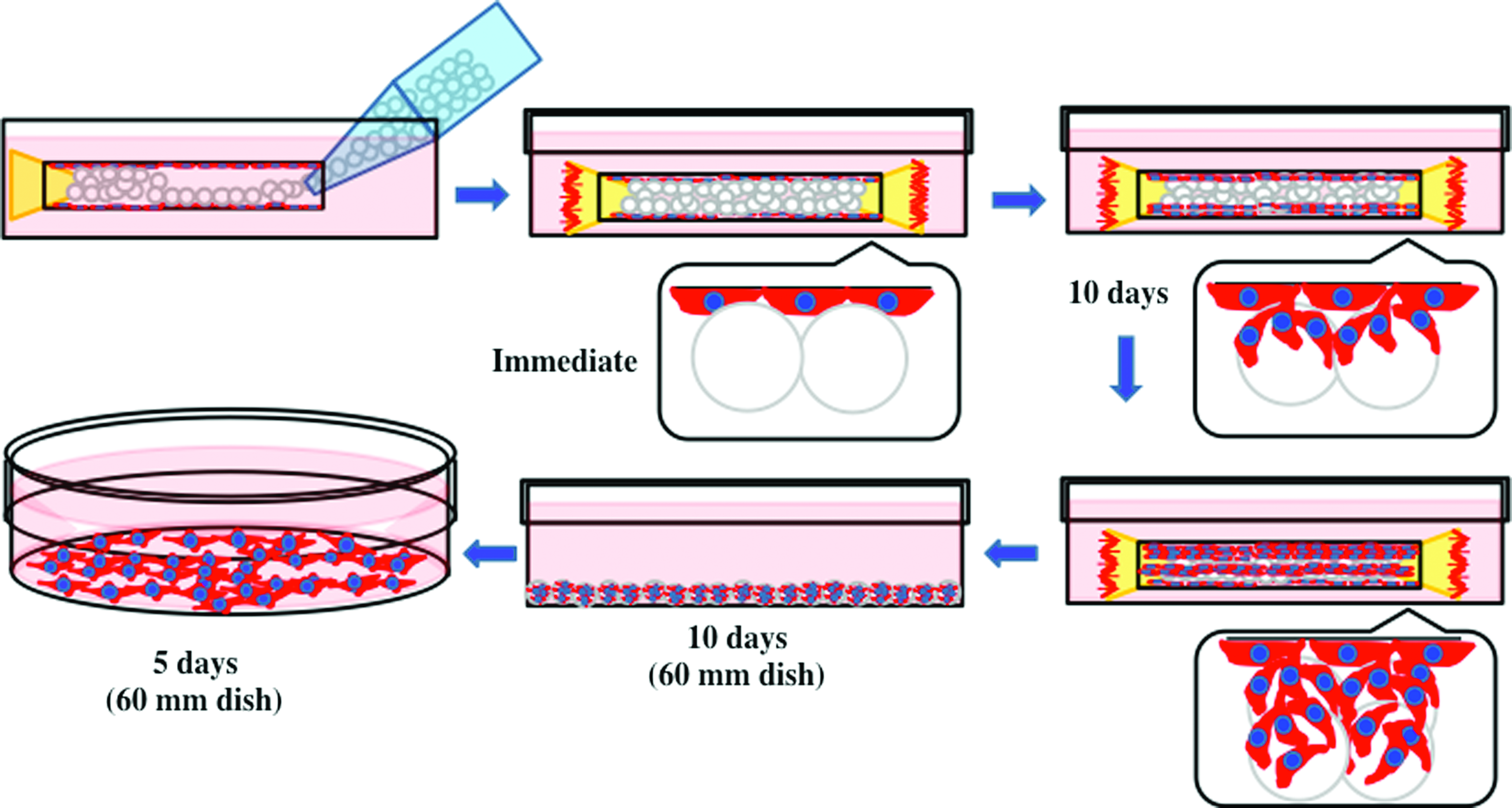
Schematic illustration of the multistep process for the isolation of primary culture of rat ECs using PCL microcarriers. Color images available online at
Cell morphology, growth, and phenotype
As predicted, a growing number of ECs migrated out of the inner wall of the blood vessel onto the surface of the scaffold over time (Fig. 5). After 5 days of culture, ECs were observed on microcarriers, although relatively few cells were present (Fig. 5A, B). However, after 10 days, many more cells were present on microcarriers (Fig. 5C, D). At this time, cells were then transferred to a 35-mm dish and cultured. ECs started to migrate from the microcarriers onto the plastic substrate of the culture dish from 2 days postincubation (Fig. 6A–C), and the culture was maintained for 10 days. The resulting cell population was then transferred to a 60-mm dish and cultured continuously for 5 days. The cell population grew in size to form a monolayer with cells displaying morphology typical of stromal cells and cell counts indicated population growth (Fig. 6D–F).
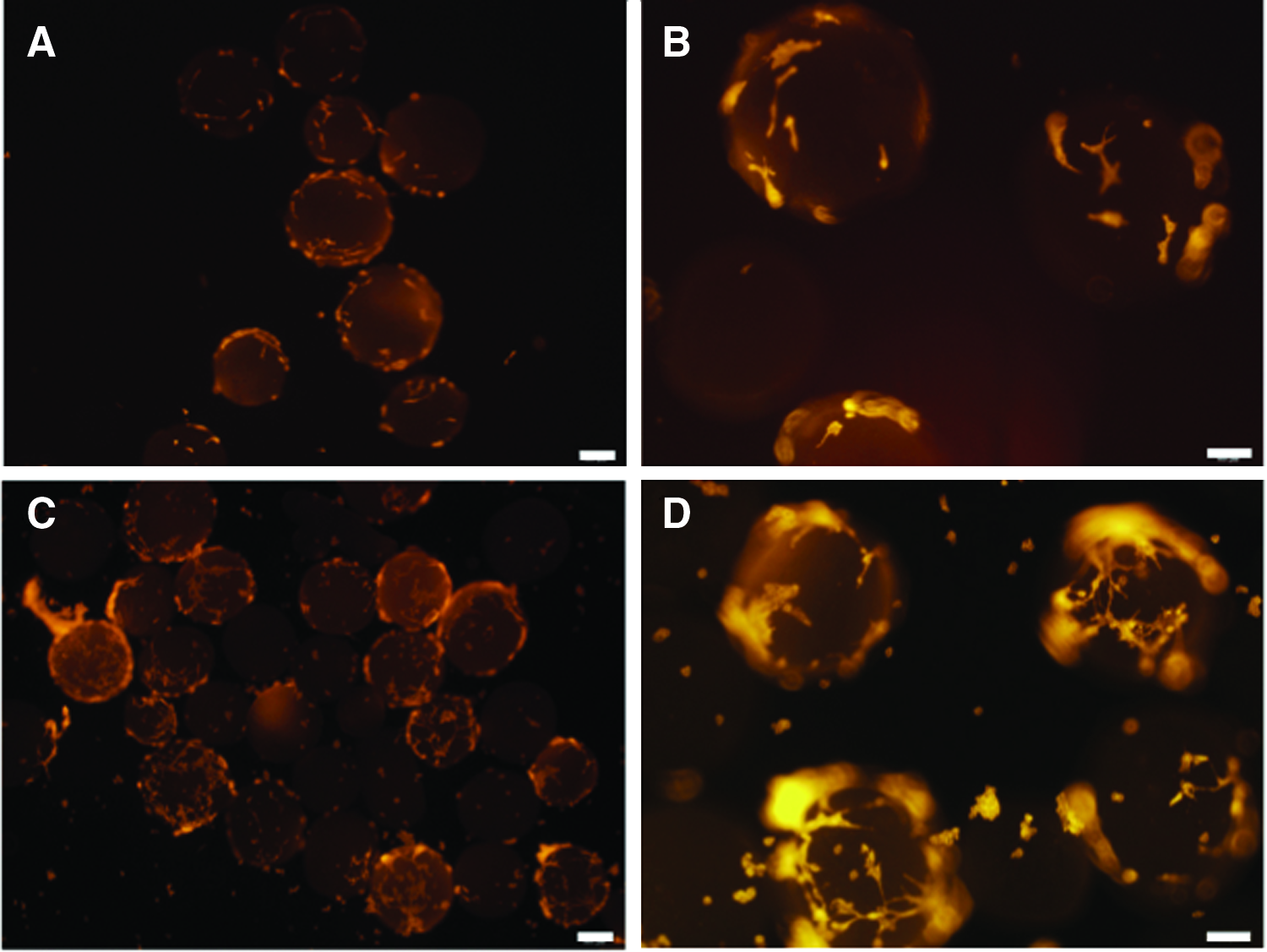
Immunofluorescent labeling to characterize EC attachment to PCL microcarriers. Cellularity after
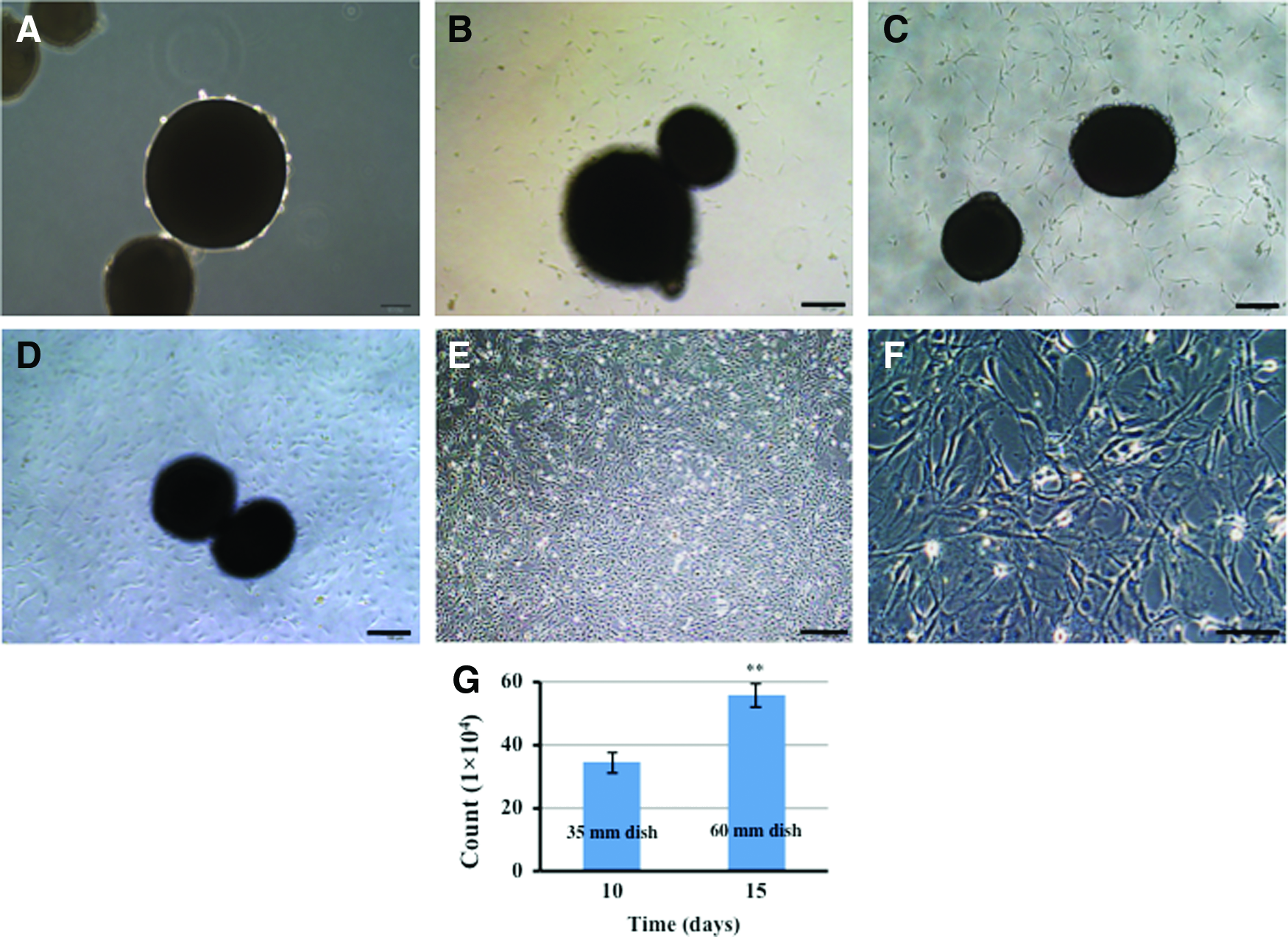
Phase-contrast micrograph images of primary aortic ECs migrating from microcarriers onto tissue culture plastic over time at 0, 2, 6, and 9 days
The functional capacity and phenotype of the cells were then assessed to determine whether the expanded cell population from microcarriers, cultured in the aorta lumen, was indeed ECs that have appropriate functional characteristics. A tubule formation assay performed on Matrigel demonstrated that the cells, when placed in the appropriate biochemical and biophysical environment, are capable of forming tubule-like structures (Fig. 7A). Moreover, the cells in culture were positive for the EC marker vWF (Fig. 7B), indicating that the tubule-like network was not an artifact of the vascular culture conditions on a non-EC type. Finally, the cell population resulting from the overall culture period was assessed for CD31 expression using flow cytometry to determine the total proportion of cells within the population that were truly ECs. Ninety-three percent of the resultant population was positive for CD31, confirming the enhanced purity, even after expansion, using the novel microcarrier-based isolation method (Fig. 7C).
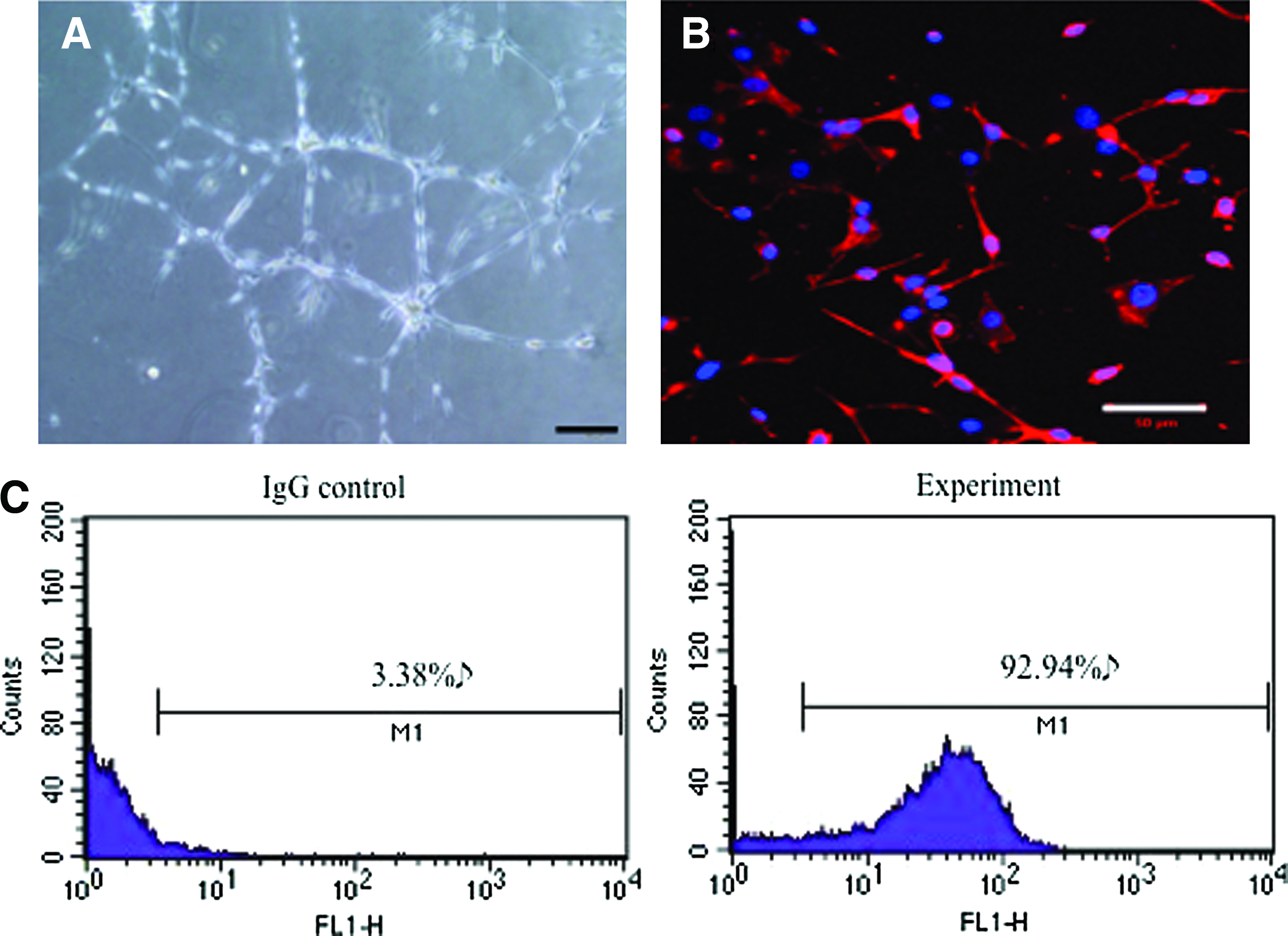
Phenotypic characterization of primary vascular ECs.
Discussion
Several methods have been routinely adopted for isolation of ECs, including enzymatic digestion, mechanical/physical methods, and outgrowth from explants. 4 Each method has advantages for isolating ECs from a variety of heterogeneously different target tissues; yet, each also brings its own limitations, with high purity often coming at the expense of yield or vice versa.
Outgrowth of ECs from tissue explants carries the risk of contamination from non-EC types present in the tissue. For example, it has been demonstrated that low-density lipoprotein presented in one of the two modified forms can induce outgrowth of either ECs or smooth muscle cells, depending on its state. 18 Our initial characterization of outgrowth cells from rat aortic tissue explants indicated that only 60% of the population obtained after expansion for 5 days in vitro was positive for the endothelial marker CD31. Therefore, we sought to assess whether PCL microcarriers could be used to isolate ECs at high purity from intact aortas under passive culture conditions.
We utilized PCL microcarriers because among the frequently used biocompatible degradable polymers, such as PCL, poly(lactic acid) (PLA), poly(glycolic acid) (PGA), and their copolymers, PCL is the least degradable among them and, hence, is thought to provide the most stable scaffold to support cells without the concern of degradation-related issues during cell isolation. However, it might be useful to study the compositional effects on cell isolation efficiency in the near future.
Due to the lumen diameter of the aortas (∼1.33±0.09 mm), we introduced large microcarriers of 200–500 μm to the vessel lumen and cultured under static conditions after prewetting the aorta and the microcarriers. The result was migration of ECs from the vessel lumen to the microcarrier surface and there was an increase in microcarrier cellularity from day 5 to 10 of incubation. Early studies isolating the microvascular endothelium with microcarriers utilized perfusion and cold buffer containing EDTA to shock ECs mediating their detachment from the basement membrane and attachment to the microcarrier surface.11,12 The extent to which this will impact on cell viability and phenotype, by selecting out a resistant population is unclear, although a cell population resilient to bioprocess forces is clearly attractive. However, our aim was to increase the purity of the population using a passive methodology, and cell-loaded microcarriers were placed in 35-mm tissue culture dishes, where cells readily migrated onto the plastic and formed a monolayer by 10 days of culture. The population could be expanded further still after passaging to a 60-mm dish, and the subsequent expanded population was more than 90% positive for CD31, expressed vWF and demonstrated functional capability to form tubule-like structures on Matrigel.
The method we have described may be suitable for isolating ECs from smaller caliber vessels, using microcarriers of smaller diameter where necessary. This would make the isolation amenable for isolating a range of topographically distinct endothelial populations from different regions such as the arteries and veins, large-diameter vessels versus small-diameter vessels. Subsequent research will focus on applying the methodology for isolating ECs from different types of vessels assessing the impact of microcarrier size.
In conclusion, we demonstrated the proof of concept that PCL microcarriers could be used as a passive means of high-purity isolation of ECs from intact aortas.
Footnotes
Disclosure Statement
No competing financial interests exist.