
Abstract
Cell–cell contact between pancreatic β-cells is important for maintaining survival and normal insulin secretion. Various techniques have been developed to promote cell–cell contact between β-cells, but a simple yet robust method that affords precise control over three-dimensional (3D) β-cell cluster size has not been demonstrated. To address this need, we developed a poly(ethylene glycol) (PEG) hydrogel microwell platform using photolithography. This microwell cell-culture platform promotes the formation of 3D β-cell aggregates of defined sizes from 25 to 210 μm in diameter. Using this platform, mouse insulinoma 6 (MIN6) β-cells formed aggregates with cell–cell adherin junctions. These naturally formed cell aggregates with controllable sizes can be removed from the microwells for macroencapsulation, implantation, or other biological assays. When removed and subsequently encapsulated in PEG hydrogels, the aggregated cell clusters demonstrated improved cellular viability (>90%) over 7 days in culture, while the β-cells encapsulated as single cells maintained only 20% viability. Aggregated MIN6 cells also exhibited more than fourfold higher insulin secretion in response to a glucose challenge compared with encapsulated single β-cells. Further, the cell aggregates stained positively for E-cadherin, indicative of the formation of cell junctions. Using this hydrogel microwell cell-culture method, viable and functional β-cell aggregates of specific sizes were created, providing a platform from which other biologically relevant questions may be answered.
Introduction
Insulin-secreting β-cells exist in complex cell clusters called the islets of Langerhans within the pancreas. β-cells have extensive contact through adherin and gap junctions with other β-cells as well as other cell types (e.g., α-, δ-, and pancreatic polypeptide cells) within islets. For in vitro culture and experimentation, islets are often isolated from the pancreas and dissociated into single cells, which results in decreased insulin secretion during glucose challenge.7–9 By reintroducing contact with other β-cells, glucose-stimulated insulin secretion was found to increase over single β-cells isolated from islets.9,10 The same has been shown for model β-cell lines, namely β-cells that are in contact with other β-cells show higher levels of insulin secretion during glucose challenge than single β-cells. 11 β-cell contact has also been implicated in cell survival when culturing in synthetic hydrogels, with cells having cell–cell contact or cell-contact mimicry displaying higher viability than cells that lack these interactions. 12
Recognizing the importance of cell–cell contact for β-cell survival and function, several methods have evolved that promote the formation of cell–cell junctions and the creation of β-cell aggregates or pseudoislets. The most common method for aggregating β-cells is cluster self-assembly in either static suspension or facilitated by culture on an orbital shaker in nonadhesive tissue culture plates.13,14 While this widely reported method requires no special equipment, it affords little control over the size and homogeneity of the aggregates, and large, irregular cell agglomerates often form in a rotational culture. Alternatively, a hanging-drop method has been used to reaggregate islets into relatively uniform spherical clusters of ∼100 μm in diameter. 15 However, this method has not shown the versatility needed to create stable aggregates of different sizes and is instead limited to a narrow size range for the aggregates.15,16 To address some of these limitations, dielectrophoresis has been used to aggregate insulinoma cells into at least two different-sized three-dimensional (3D) constructs. Radio frequency voltages applied to specific electrodes generate dielectrophorectic forces, which, when properly matched to the cell membrane capacitance and conductance, can be used to condense a single-cell suspension of β-cells into cell clusters of defined sizes. 16 This method affords tight control over cluster size and the ability to incorporate labeled nanospheres, but has not shown the facile tuning of aggregate size, and dielectrophoresis requires specific equipment that may not be available in many labs. More recently, microcontact printing has been used for the creation of β-cell aggregates. In one example, different size spots of the cell-adhesive protein laminin were printed on aldehyde-terminated glass coverslips that form multi-cellular aggregates of β-cells of different sizes. 17 Microcontact printing allows the manipulation of aggregate size by changing the size of the printed area, but the cell clusters could not be removed from the scaffolds for further biological tests or implantation, and the clusters were only two to three cell layers in thickness. 17
Complementary to microcontact printing, microwell arrays can be quickly fabricated using photolithography that creates miniaturized cell-culture wells and forms 3D cell aggregates with dimensions which scale with well size. Devices are readily constructed with well depths and diameters ranging from tens to hundreds of microns. One challenge, however, is the efficient seeding of cells in a large number of microwells. Several groups have shown the utility of these devices to control cell aggregation for many cell types, such as ESCs and fibroblasts. Many polymeric materials have been utilized for microwell cell culture, including polyesters, 18 polyurethanes, 19 and poly(ethylene glycol)s (PEGs).3,4,19–23
PEG provides particular advantages for microwell fabrication due to its bioinertness, ability to tune the device material properties based on the PEG molecular weight, and success in many microwell cell-culture platforms. For example, PEG microwell devices have been used to direct ESC differentiation by creating embryoid bodies of defined sizes3,20,21 or different concavities. 22 Conversely, 3D PEG microwell arrays can also be used to maintain an undifferentiated phenotype of ESCs for several weeks in culture. 4 PEG microwell arrays functionalized with collagen were also found to direct 3T3 fibroblasts and primary hepatocyte cells attachment and isolation, 23 and PEG-fibrinogen microwell arrays maintained the liver-specific function of hepatocytes grown in 3D culture. 24
The broad applicability of these devices and the facile fabrication shown in this work make microlithographic devices particularly attractive as a tool for reaggregation of primary β-cells or β-cell lines. In this contribution, we aimed at using a contact-lithographic process 25 that creates PEG-based microwell devices which would enable the formation of uniformly sized aggregates of the immortalized β-cell line, mouse insulinoma 6 (MIN6). We sought to exploit the bioinert and nonadhesive properties of PEG to create devices that would promote cell–cell aggregation in microwells, allow culture for several days, the subsequent removal of the β-cell aggregates, and encapsulation for further studies. These MIN6 aggregates were then compared with encapsulated MIN6 single cells to assess cellular viability, metabolic activity, and the ability to secrete insulin. Immunofluorescent staining was used to examine the formation of cell–cell junctions between aggregated MIN6, as well as the maintenance of intracellular insulin content.
Materials and Methods
PEG microwell synthesis
Poly(ethylene glycol) diacrylate (PEGDA) (Mn≈3000 Da) was synthesized from hydroxy-terminated PEG (Sigma) as previously described. 26 Microwell devices were created using contact photolithography (Fig. 1) as previously outlined.25,27 Briefly, a prepolymer precursor solution was mixed containing 15 total wt% macromer (75 mol% PEG-monoacrylate [PEGA, Mn≈400 Da; Monomer-Polymer and Dajac Labs], and 25 mol% PEGDA [Mn≈3000 Da]), 0.5 wt% 4-(2-hydroxyethoxy)phenyl-(2-hydroxy-2-propyl)ketone (Irgacure 2959; Ciba), and 84.5 wt% Hank's balanced salt solution (HBSS; Gibco). For fluorescent imaging, some prepolymer solutions contained ∼300 μM methacryloxyethyl thiocarbonyl Rhodamine B (Polysciences, Inc.) The solution was pipetted between an acrylated glass slide and a chrome photomask (Photo Sciences, Inc.) separated by plastic spacers with a defined thickness (Artus, Inc.) (Fig. 1A). The acrylation of glass slides was achieved via chemical vapor deposition of (3-acryloxypropyl)-trimethoxysilane (Gelest) on piranha solution (3:1 sulfuric acid/hydrogen peroxide) cleaned glass surfaces 28 before polymerization. Prepolymer solutions were irradiated using an Oriel Flood Exposure system with a mercury arc lamp and dichroic mirror (λ=350–450 nm, 350 W; Oriel Instruments) through a chrome photomask (patterned squares 50, 100, 150, 200, and 300 μm) for 60 s to form crosslinked hydrogels. The polymerized microwell devices were incubated in deionized water to remove unreacted monomer and subsequently sterilized in 70% ethanol. The devices were stored in HBSS supplemented with 1% Penstrep (Gibco) and 0.2% Fungizone (Gibco) under a UV germicidal lamp (Fisher; G64T5L-CB) overnight to complete the sterilization.
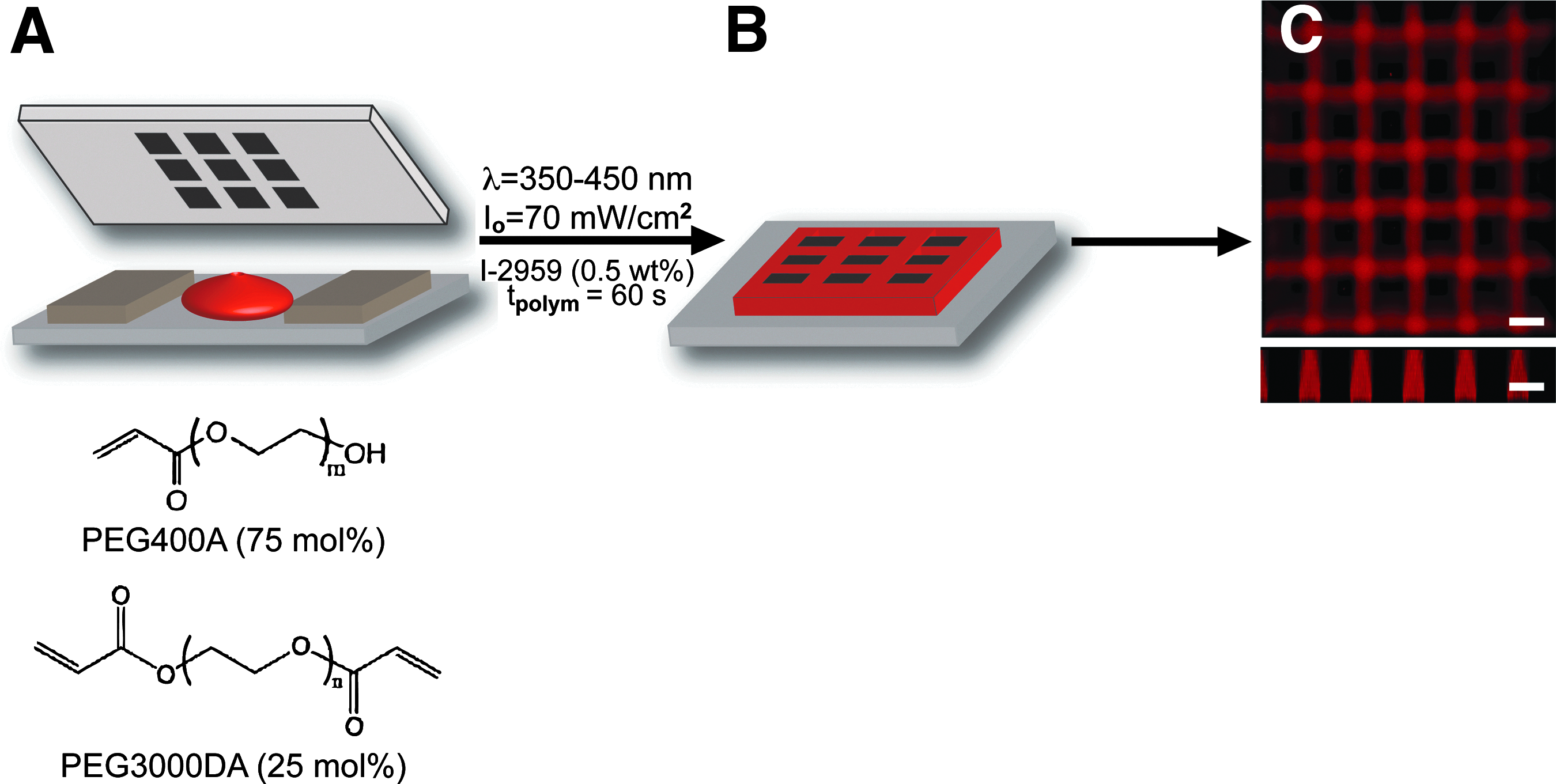
Formation of hydrogel microwell devices.
Cell culture and seeding
MIN6 cells were maintained in low-glucose Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (Gibco), 1% PenStrep, and 0.2% Fungizone. Cells were detached from culture surfaces using 0.05% trypsin/ethylenediaminetetraacetic acid, dissociated into single cells, and seeded on microwell devices at 3×106 cells/mL using a centrifugation-assisted method (1200 rpm for 2.5 min). 22 After centrifugation, the cells and devices were incubated on an orbital shaker (35 rpm, 5% CO2, 37°C) for at least 2 h. The seeding process was repeated once for a total of two seeding events for each device. The cells were cultured under static conditions in the incubator for 5 days, changing media every 2–3 days. After 5 days of culture, the cell clusters were either imaged or removed from the devices and encapsulated for further studies.
Confocal imaging and image analysis
Multicellular aggregates and microwell devices were imaged using a Zeiss LSM 710 NLO confocal microscope (Carl Zeiss). To measure the cell aggregates, cells were stained with 10 μM CellTracker™ Green 5-chloromethylfluorecein diacetate (Invitrogen) as per the manufacturer's instructions before seeding. Z-stack images were collected, and aggregate diameter was quantified using ImageJ (National Institutes of Health). Briefly, the image stacks were compressed to one plane, made binary, and evaluated using the ellipse tool in ImageJ; the major diameter of the bestfit ellipse was used as the aggregate diameter. A total of at least 250 aggregates from at least 10 devices seeded in a minimum of three separate experiments were measured for each well dimension.
The average number of cells per aggregate was determined after 5 days of aggregation in the microwell devices. The aggregates were removed from microwell devices and dissociated into a single-cell suspension using trypsin. The number of single cells was counted using a hemocytometer, and the average number of cells per aggregate of different sizes was calculated.
Aggregate removal and encapsulation
PEGDA (Mn≈10,000 Da) and lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) photoinitiator were synthesized as previously described.26,29 Cell aggregates were removed from microwell devices by gently rinsing with media and collected in 50 mL centrifuge tubes. Aggregates were allowed to settle, and the supernatant was removed. The aggregates were then gently resuspended using wide-bore pipettes in the encapsulation prepolymer solution consisting of 10 wt% PEGDA (Mn≈10,000 Da), 0.05 wt% LAP, and HBSS. 30 μL of the prepolymer cell suspension were polymerized in 1 mL syringe tips under 352 nm light in a sterile hood for 3 min. Similar photoencapsulation methods have been reported in applications using both β-cell lines12,14,30 and primary islets.12,31–33 The gels were immediately removed to low-glucose DMEM, and the unreacted prepolymer solution was allowed to diffuse out of the hydrogels overnight.
Cell viability
Cells were encapsulated as either aggregates or single cells at a cell density of 1×106 cells/mL for viability assessment. Cells were stained for viability on days 1, 3, and 7 after encapsulation using a LIVE/DEAD Viability/Cytotoxicity Kit for mammalian cells (Invitrogen) (calcein AM at 2 μM, ethidium homodimer-1 at 4 μM) and visualized using confocal microscopy where live cells stain green and DNA of dead cells stains red. The metabolic activity for encapsulated single cells at a cell density of 2×106 cells/mL and cell aggregates was measured using AlamarBlue (Invitrogen). Samples were incubated in cell-culture media containing 10% of the AlamarBlue reagent for 16 h, and the fluorescent signal of the supernatant (excitation: 560 nm, emission: 590 nm) corresponds to the metabolic activity of the encapsulated cells within the gels. Fluorescence of the collected samples was normalized to that recorded on day 0, yielding a percent of metabolically active cells.
Immunofluorescent staining
Encapsulated MIN6 aggregates were immunofluorescently labeled to determine the presence of intracellular insulin and cell–cell junctions indicated by the presentation of organized E-cadherin. The cells were fixed 2–4 days after encapsulation in 4% paraformaldehyde for 15 min at room temperature, rinsed with phosphate-buffered saline (PBS), and blocked with bovine serum albumin (BSA; Sigma) to prevent nonspecific staining. The cells were incubated in a guinea pig polyclonal antiinsulin primary antibody (1:50, Abcam-ab7842) and a mouse monoclonal antibody against E-cadherin (1:150; BD Transduction - #610181) overnight at 4°C to allow sufficient diffusion through the gel. The gels were rinsed in PBS containing 3% BSA and then incubated in appropriate secondary antibodies (Alexa Fluor 488 donkey-anti-mouse IgG and Alexa Fluor 633 goat-anti-guinea pig IgG, 10 μg/mL; Invitrogen) overnight at 4°C. The gels were rinsed again and then incubated in 11 μM 4'-6-diamidino-2-phenylindole (DAPI, Invitrogen) to counterstain the nuclei. Encapsulated aggregates were imaged using a Zeiss NLO 710 laser scanning confocal microscope.
Insulin secretion
Cells were encapsulated as either aggregates or single cells maintaining the same initial number of cells per gel. The number of aggregates per gel was determined postencapsulation by counting using an inverted microscope (Nikon Eclipse TE 2000), and the total number of cells per gel was calculated. A complementary number of single cells were then encapsulated in separate gels for comparison. Gels containing either single cells or aggregates were subjected to a glucose challenge test 2 days after encapsulation, and the secreted insulin was measured via insulin enzyme-linked immunosorbent assay (ELISA). Briefly, the cells were conditioned in low (2 mM)-glucose Kreb-Ringer Buffer (KRB) for 45 min. The cells were then incubated in high-glucose KRB (25 mM) for 1 h, and the amount of insulin secreted was measured using a mouse insulin sandwich ELISA kit (Mercodia). The secreted insulin was normalized to the initial number of cells encapsulated in each gel.
Statistical analysis
Due to the large number of data points analyzed, average aggregate diameters are shown as the numerical mean, and the error bars represent a standard deviation from the mean. All other data are reported as the mean with error bars indicating the standard error of the mean. Statistical significance was determined using a one-way analysis of variance (ANOVA) followed by Tukey's test for the aggregate diameters and using a one-tailed Student's t-test for insulin secretion. Values were considered significant if p<0.05, unless otherwise noted.
Results
Cell–cell interactions between pancreatic β-cells have been shown to be extremely important for their function and viability both in vitro and in vivo.7–10 As a result, several parameters should be considered when designing a culture platform that promotes cell–cell contact for β-cells while maintaining their utility for transplantation. A material platform that supports cell–cell contact over cell-material interactions will facilitate the formation of β-cell aggregates. Additionally, the material platform should be cytocompatible with the β-cells for promoting long-term cellular viability and ideally allow control over the cell aggregate size. Finally, it should also allow for removal of the cellular aggregates from the in vitro culture platform and transfer to an in vivo compatible system for transplantation.
Formation of PEG microwell arrays using photolithography
Microwell arrays were fabricated using PEG monomers which create hydrogels that promote MIN6 aggregate formation. Specifically, a formulation containing 75 mol% PEGA (Mn≈400 Da) and 25 mol% PEGDA (Mn≈3000 Da) at 15 wt% of the total solution were found to produce robust gels while minimizing dimensional changes after polymerization. Figure 1 shows a schematic of the microwell formation, as well as optical images of polymerized hydrogel devices. Using standard photolithographic masks, well diameters were varied from 50 to 300 μm with well depths of 100 μm set by plastic shims inserted between the photomask and glass surface (Fig. 1A). The hydrogel cross-linking and microwell formation, as well as attachment to the acrylated glass surface, were achieved within 60 s of polymerization with 70 mW/cm2 of 350–450 nm ultraviolet light. These conditions yielded adequate polymerization time as measured by in situ rheometry (Supplementary Fig. S1; Supplementary Data are available online at
MIN6 aggregate uniformly in 3D PEG microwell arrays
Dispersed single MIN6 cells suspended at a density of 3×106 cells/mL were seeded into square PEG microwell devices via forced centrifugation. Well dimensions on the devices had a depth of 100 μm and widths ranging from 50 to 300 μm. Seeded cells formed tight aggregates naturally within 5 days of in vitro culture. Figure 2 shows representative images of MIN6 cellular aggregates formed in microwells with widths measuring 100 μm (A), 200 μm (B), and 300 μm (C). Cell aggregates removed from microwell devices with respective well dimensions are shown in Figure 2D–F. A high percentage of the microwells were filled, and the MIN6 cells clustered together to form 3D aggregates reproducibly that were relatively uniform in size and with dimensions that scaled with the microdevice well dimensions (Fig. 2A–C). After 5 days of culture, the MIN6 cells formed sufficient cell–cell junctions to maintain their size and shape during removal of the aggregates from the microwell devices (Fig. 2D–F).
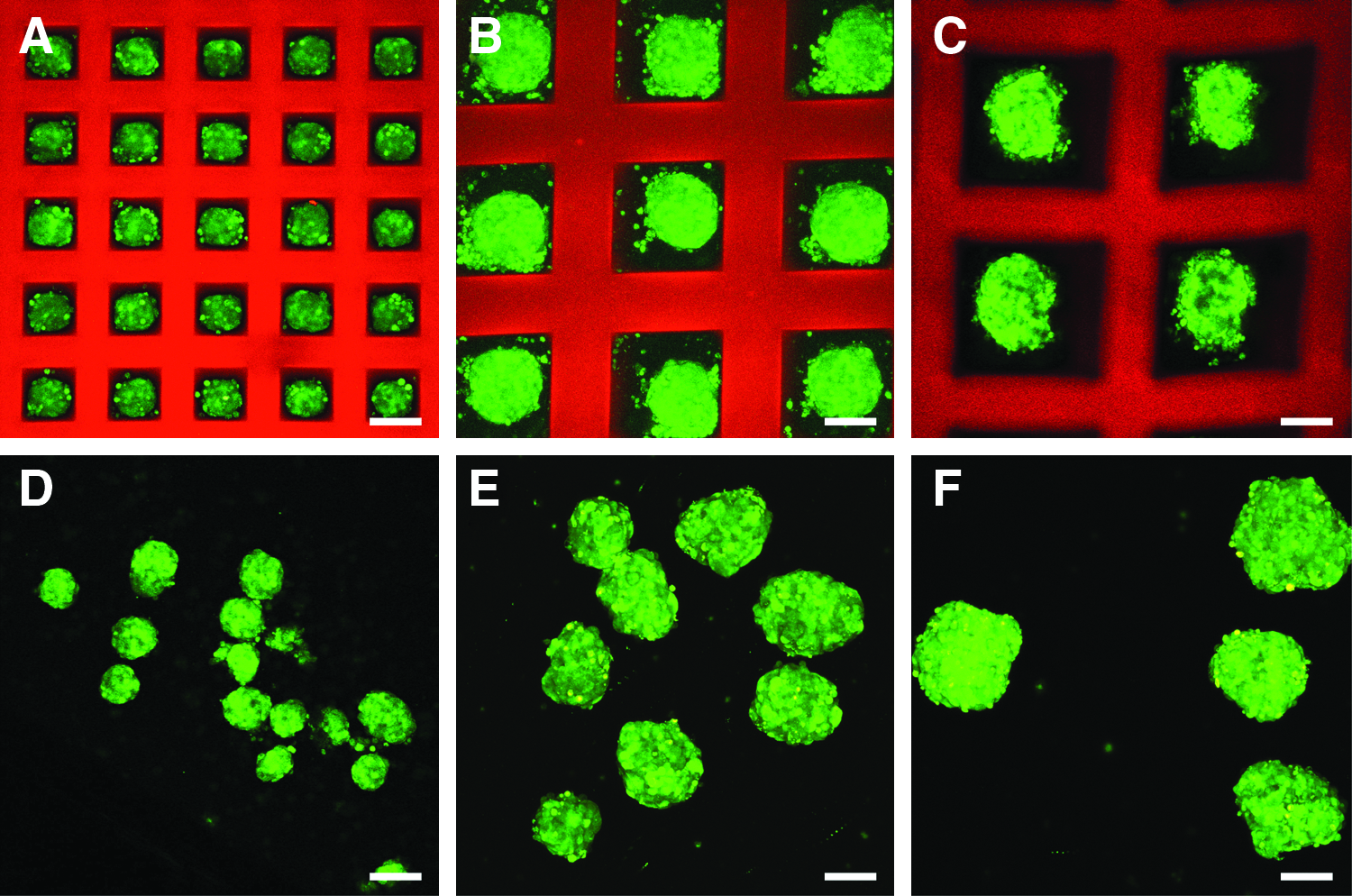
Aggregated mouse insulinoma 6 (MIN6) cell clusters in poly(ethylene glycol) (PEG) microwell devices. MIN6 cells were suspended at a density of 3×106 cells/mL and seeded within the microwell devices by centrifugation at 1200 rpm for 2.5 min. MIN6 cells were stained green with CellTracker™ Green 5-chloromethylfluorecein diacetate before seeding. Confocal images were taken 5 days after seeding
To assess the uniformity and scalability of MIN6 cell aggregates formed using hydrogel microwell arrays in a more quantitative manner, the aggregate diameter was measured using confocal laser scanning microscopy and image analysis via Image J. The average aggregate diameters were found to depend on the cross-sectional area of the square microwells ranging in width dimensions from 50 to 300 μm, and all the average aggregate diameters were statistically significant from each other with p<0.01 using a one-way ANOVA followed by Tukey's test (Fig. 3A). Wells with a width of 50 μm (w50), 100 μm (w100), 150 μm (w150), 200 μm (w200), and 300 μm (w300) yielded cell aggregates with an average diameter of 25±3, 80±9, 120±10, 160±20, and 210±30 μm, respectively. To test the reproducibility of MIN6 aggregate formation using these PEG microwell devices, aggregates from at least 10 different microwell devices seeded in at least 3 separate experiments were measured, and the individual diameter measurements were plotted versus microwell width to show the size distribution of aggregates formed after 5 days of culture (Fig. 3B). Using this method, it was possible to create a large number of MIN6 aggregates with a narrow distribution in the aggregate dimensions; however, the distribution in the aggregate diameter became broader as the cross-sectional area of the well increased.
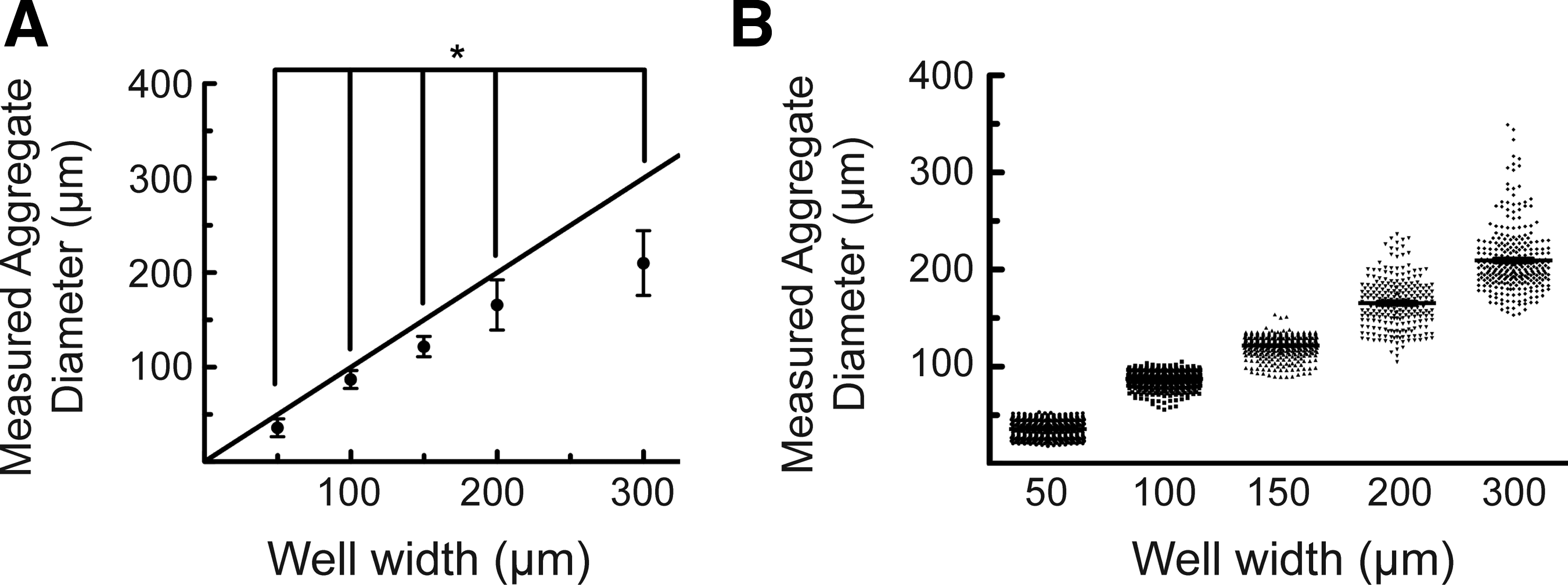
Characterization of MIN6 aggregate size as a function of microwell dimension.
Beyond aggregate size and homogeneity, it was also important to estimate the number of cells contained within the aggregates of different sizes in order to normalize the assay results to cell number, as well as to compare the cell aggregate data with that of single cells. As expected, the total number of cells per aggregate was found to increase with well volume and a corresponding increase in the aggregate diameter. Specifically, aggregates from devices fabricated with well dimensions of 100, 200, and 300 μm edges contained 490±20, 780±120, and 2400±190 cells, respectively, at the time of encapsulation (Fig. 4K).
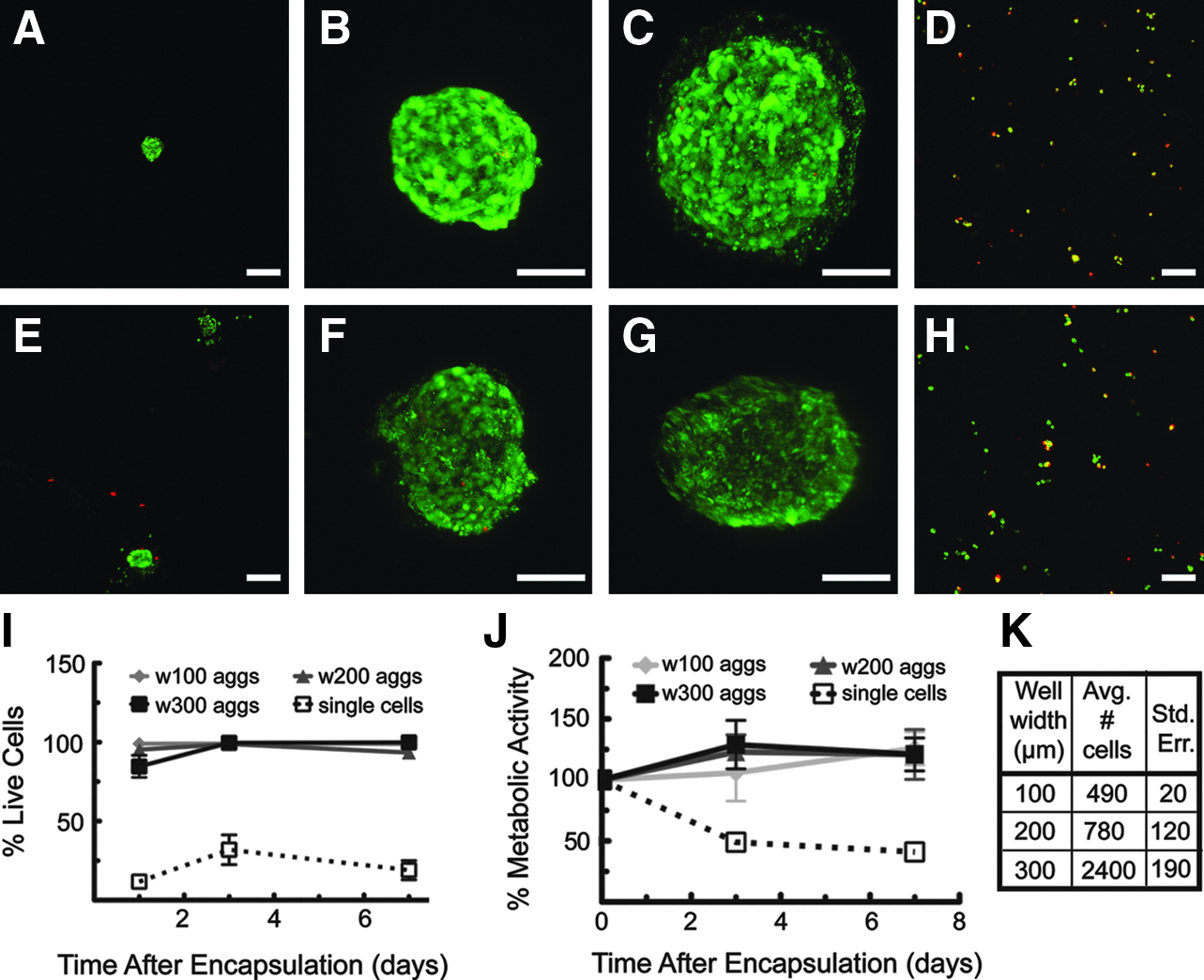
LIVE/DEAD® images of encapsulated MIN6 aggregates and single cells
MIN6 cells show increased postencapsulation viability and metabolic activity when cultured as aggregates
The survival of MIN6 cells encapsulated in PEGDA capsules was determined for both single cells and aggregates removed from devices using a LIVE/DEAD viability staining kit. Aggregated MIN6 cells maintained high levels of viability throughout the experiment (>90%), including cell seeding, culture within and removal from the PEG microwells, and encapsulation in PEGDA hydrogels. MIN6 aggregates show improved viability compared with single cells encapsulated at the same number of cells per gel over 7 days in culture (Fig. 4). MIN6 single cells encapsulated at a cell density of 1×106 cells/mL showed an average of 30% live cells on day 3 and 20% live cells on day 7, while>90% of aggregated MIN6 cells stained viable on days 3 and 7 (Fig. 4A–I). The loss of single MIN6 viability is likely due to anoikis 33 and not from the photoencapsulation method, as this and similar photoencapsulation schemes have been previously used with MIN6 cells successfully.14,30,33,34 As a further measure of cell viability, metabolic activity was monitored and found to decrease by 50% on day 3 and 60% on day 7 for encapsulated MIN6 single cells at a density of 2×106 cells/mL. In contrast, encapsulated MIN6 aggregates did not show a decrease in metabolic activity over the entire week of culture (Fig. 4J). Collectively, these results indicate that MIN6 cells show improved levels of viability and metabolic activity when aggregated before encapsulation compared with single cells encapsulated at the same number of cells per gel.
Aggregated MIN6 maintain expression of intracellular insulin and E-cadherin
To test whether MIN6 cells maintained their insulin content throughout the seeding, aggregation, and encapsulation processes, encapsulated aggregates were stained using a polyclonal antibody directed against murine insulin. Aggregated MIN6 cells stained strongly positive for insulin content, indicating that the cells were still capable of producing insulin after in vitro culture in microwell devices (Fig. 5B, E, H). Aggregated β-cells also expressed the intercellular binding protein E-cadherin (Fig. 5C, F, I), indicating cell–cell junctions. An overlay of the immunostaining images is included in Supplementary Figure S2. MIN6 cell clusters were not fluorescent when stained with secondary antibody alone (data not shown).
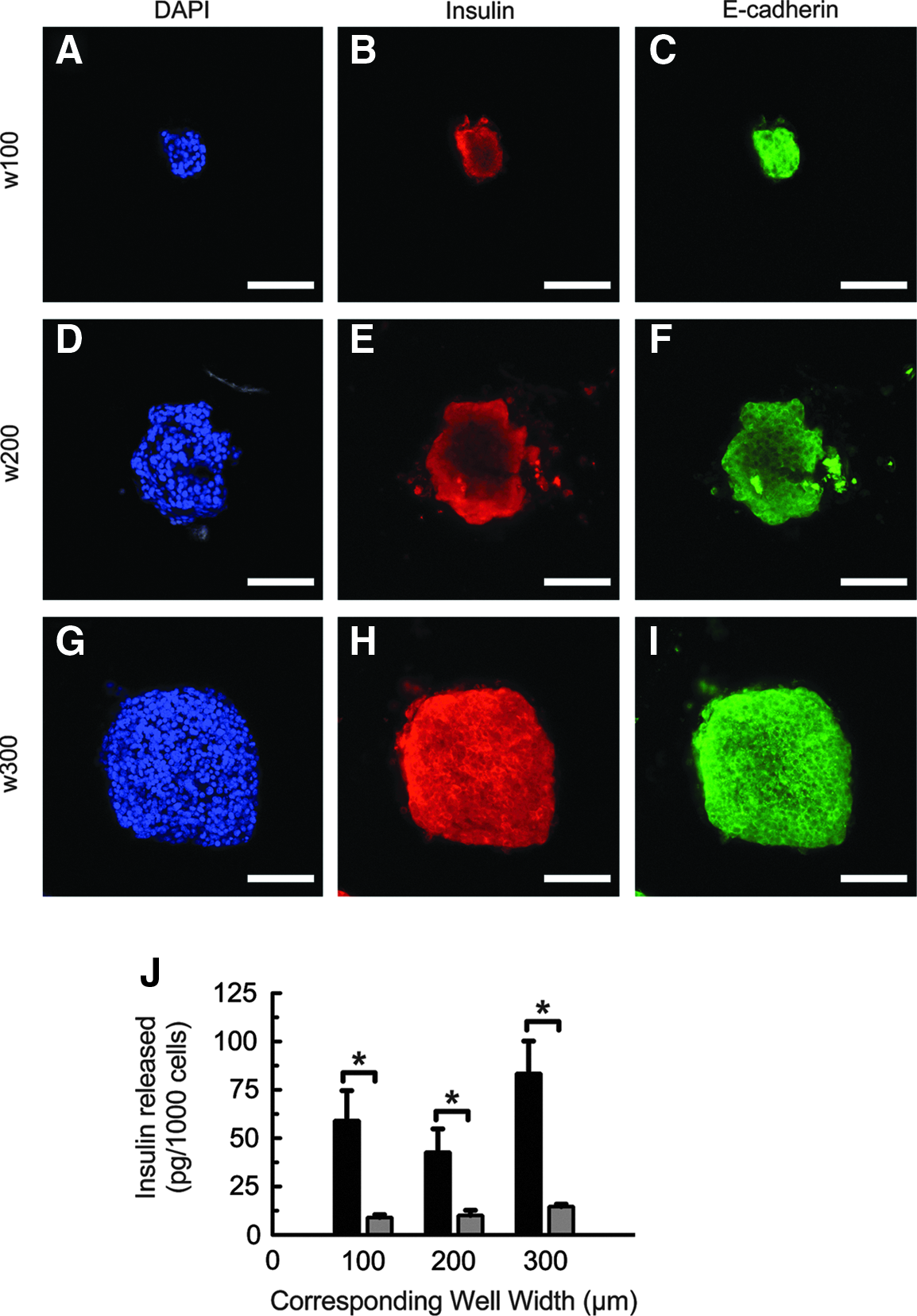
Functional expression of MIN6 aggregates.
Aggregated MIN6 cells show increased insulin secretion compared with single cells
To test for stimulated insulin secretion, encapsulated MIN6 single cells and aggregates were first conditioned in KRB containing low glucose (2 mM) and then exposed to KRB containing high glucose (25 mM). Figure 5J shows insulin secretion from MIN6 single cells compared with aggregates 2 days after encapsulation. The amount of insulin secreted from the cells was measured using a sandwich ELISA and then normalized to the total number of cells in the gel. Compared with an equal number of single cells, aggregated MIN6 cells were found to secrete more than fourfold the amount of insulin on a per-cell basis (Fig. 5J), and per-cell insulin release from MIN6 cells was not found to depend on aggregate size. A comparison of insulin secreted from aggregated and single MIN6 cells normalized to cellular adenosine triphosphate (ATP) is shown in Supplementary Figure S3.
Discussion
Isolated cells from pancreatic islets and immortalized β-cell lines require contact with other cells or basement membrane proteins—or mimicry of these contacts—to maintain function and viability when cultured in synthetic in vitro systems. Recognizing this requirement, different approaches have been developed that encourage or increase cell–cell contact for β-cells, especially before cell encapsulation.13–16 In this work, we present a method for controlling the aggregation of MIN6 cells into well-defined cluster sizes using PEG microwell devices formed by standard contact photolithographic techniques. The bioinertness of PEG provides a noncytotoxic environment that can be easily manipulated, sterilized, and used for cell culture. PEG has been shown to resist protein adhesion and cell attachment,35–38 and this characteristic not only promotes cell–cell interactions over cell-material interactions, but also allows the facile removal of cell aggregates from microwell devices.
The contact photolithographic formation of PEG microwell devices for β-cell culture shown here offers some key advantages over other microwell array formation techniques, such as soft lithography—which uses silicon masters and poly(dimethyl-siloxane), stamps to form microwell arrays. One benefit of contact lithography is the ability to easily define and change well dimensions. Wells of different cross-sectional areas require the use of separate photomasks, but the height of the device (depth of the wells) can be changed easily by varying the amount of prepolymer solution used and spacers to control the distance between the substrate and the photomask. In order to make wells of different dimensions using soft lithography, new masters and stamps should be created, which can be time consuming and costly. While the PEGDA/PEGA system shown in this work provides a robust material platform for the formation of microwells to promote defined β-cell aggregation, the nature of the copolymerization between acrylates and diacrylates for the formation of microwells is limited in scope; namely, the current formulation cannot be used to form microwells that exceed a 1:1 (width:height) well aspect ratio. If this aspect ratio is exceeded (e.g., the formation of wells that are deeper than they are wide), polymerization can occur in the dark region and result in well occlusion (Supplementary Fig. S4). This occlusion is also observed during over-polymerization, when exposing the prepolymer solution to excessive amounts of ultraviolet (UV) light. This issue is easily addressed by limiting the well height and UV exposure time during polymerization. If deeper wells are necessary, then other techniques not investigated here, such as soft lithography, might be used for well formation.
PEG microwell devices were found to lead to the formation of uniform β-cell aggregates from a single-cell suspension of MIN6 cells without the use of complex equipment. Cylindrical and cuboidal microwells were tested for 3D MIN6 cell culture, and both systems facilitated the formation of rounded cellular aggregates (Supplementary Fig. S5). Arrays of cuboidal wells were chosen for this application, as they increased the space between the material and cells, created when the cells form rounded aggregates in square spaces, allowed the flow of media to more easily remove aggregates from the microwell devices. The aggregates were readily removed from the devices and maintained their size and shape (Fig. 2), presumably due to strong cell–cell attachment and low cell-material adhesion. For subsequent encapsulation studies and/or testing transplantation studies, it is critical to be able to remove the aggregates from the devices. After removal from the microwells, the β-cell aggregates can be used for other biological experiments or encapsulated within other permissive biomaterials (e.g., PEGs 32 or alginates39,40) for implantation.
The use of PEG microwell arrays for β-cell aggregation yielded relatively uniform cell aggregates—demonstrated by narrow distributions of both aggregate diameter and cells per aggregate—which scaled in size with microwell dimension. As the microwell width was increased from 50 to 300 μm, a corresponding increase in aggregate diameter was measured (Fig. 3). As expected, the average aggregate diameter was less than the well width due to the penchant of β-cells to form clustered structures and the dominance of cell–cell interactions over cell material interactions. The range of aggregate sizes created via this method correlates well to that observed in naturally occurring murine islets,41,42 thus providing a culture platform from which the effect of aggregate size on physiologically relevant properties could be investigated. While the model β-cell line used in this work did not show a size-dependent effect on cellular viability or functional expression, but rather demonstrated exceptional viability and function over all size aggregates, the effect of islet size on viability and function has been documented41,43 suggesting that using this method to control islet reaggregation might be beneficial. In the future, we plan to use these microwell devices to reaggregate beta cells isolated from primary islets and investigate the impact that distinct pseudo-islet size has on viability and function.
It is important for long-term MIN6 culture, as well as future implantation studies, that high levels of cell survival be achieved using synthetic hydrogel encapsulation materials. This method of β-cell aggregation promotes high viability of cells when encapsulated in a synthetic hydrogel system without the need to incorporate additional matrix proteins or adhesive peptide sequences. When compared with single β-cells encapsulated at the same number of cells per gel, the aggregated MIN6 show much higher viability (over 90%) and maintain high levels of metabolic activity (Fig. 4). We attribute the loss of viability of single β-cells to lack of matrix interactions, as well as cell–cell interactions, both of which have been widely reported as affecting the survival of encapsulated β-cells.14,30,33 However, several complexities should be acknowledged when interpreting these results.
The first relates to central necrosis in cell aggregates. Although often seen during the culture of intact islets, the reaggregated β-cells did not show signs of central necrosis, as the cell-permeable viability dye was able to penetrate to the interior of the aggregates and the central cells stained green. While it is possible that the peripheral cells of the aggregates might experience a different environment that could lead to cell death, as seen with the single cells, this was not observed in the current study.In addition, while proliferation was not directly measured, it is unlikely that cell doubling contributed significantly to these results. An appreciable increase in cell density was not observed between single cells imaged 3 days after encapsulation (Fig. 4D) and 7 days after encapsulation (Fig. 4H). While not quantified, cell aggregates appeared to remain similar in size throughout the culture period within the microwells (Fig. 2A–C), after removal (Fig. 2D–F), 3 days after encapsulation (Fig. 4A–C), and 7 days after encapsulation (Fig. 4E–G), indicating only modest increases in cell number compared with the initial number of cells within the aggregate.
The maintenance of functional expression throughout cell manipulation is important for β-cells. MIN6 cells aggregated using PEG microwell arrays were shown to maintain their functional expression throughout seeding, aggregation, culture, and encapsulation. The cells preserved intracellular insulin content at high levels as evidenced by immunostaining. MIN6 cells interact with one another and maintain normal insulin release through cell–cell contact mediated, in part, by E-cadherin. 44 MIN6 cells will stain positive for aligned E-cadherin between neighboring cells when proper cell–cell contact is achieved. After aggregation, the cells stained positive for E-cadherin, which appears to be aligned at cell–cell junctions similar to cells found in intact, isolated islets (Fig. 5). This staining indicates that MIN6 cells formed proper cell–cell junctions when aggregated using PEG microwell devices, which may suggest strong cell–cell contact and communication, leading to improved viability and insulin response.
Insulin secretion in response to glucose is one of the hallmark traits of functional β-cells and should not be negatively affected by in vitro manipulation of β-cells through culture, aggregation, or encapsulation. The amount of insulin secreted by aggregated MIN6 cells is much higher than that secreted by encapsulated single cells. When the same numbers of cells are tested, the aggregated cells were found to secrete fourfold more insulin. This characteristic allows greater insulin secretion using a smaller number of cells. Since cell number is often the limiting factor in successful cell transplantation, improving the function of β-cells without increasing the required number of delivered cells is highly desirable.
In conclusion, using a PEG-based hydrogel microwell array platform, aggregates of MIN6 β-cells can be reproducibly formed. The aggregate size and average number of cells per aggregate correlated with the microwell dimensions. Aggregates could be rapidly removed from the microwell devices and encapsulated for further biological assays and applications. Encapsulated MIN6 cell aggregates maintained higher levels of viability and secreted more insulin than encapsulated single cells. The aggregated cells also stained positive for E-cadherin, indicating the formation of cell–cell junctions that are critical for enhanced insulin secretion.
Footnotes
Acknowledgments
The authors wish to acknowledge Kelly Trowbridge and Mark Tibbitt for technical assistance and helpful discussions. They gratefully acknowledge financial support from the National Institute of Health (RO1DK076084), the Howard Hughes Medical Institute, and the Department of Education's Graduate Assistance in Areas of National Need Fellowship to ABB.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.