
Abstract
Odontogenesis, the intricate process of tooth development, involves complex interactions between oral ectoderm epithelial cells and ectomesenchymal cells derived from the cephalic neural crest, regulated by major signaling pathways. Dental developmental anomalies provide valuable insights for the clinical diagnosis of rare diseases. More than 30% of patients with rare diseases who undergo molecular analysis suffer from diagnostic errancy. In the search for up-to-date technologies and methods to study the pathophysiology of new candidate genetic variants, causing tooth mineralized tissue anomalies, we have developed an original model of tooth organoids with human or mouse cell lines of ameloblast-like cells and odontoblasts derived from the pulp. This in vitro 3D cellular model reproducing the two main compartments of the bell stage of tooth development between ameloblasts and odontoblasts, specific to enamel and dentin morphogenesis, respectively, mimics the epithelial–mesenchymal interactions during the dental bell stage of tooth morphogenesis and will facilitate the study of enamel and dentin genetic anomalies, allowing the functional validation of newly identified mutations (variants of uncertain significance or new candidate genes).
Impact Statement
We have developed and characterized innovative 3D bilayer organotypic cellular models with ameloblast-like cells and odontoblast-like cells (both murine and human) reproducing some of the interactions during the bell stage of odontogenesis. This is a first model of such type, considering human lineages and not only primary dental cells, with high reproducibility, and including recently developed polymeric poly-(lactic-coglycolic acid) microscaffolds for increasing cell survival and favoring cell differentiation. This work opens new perspectives to the study of tooth morphogenesis in vitro and associated genetic disorders, either for fundamental studies or for the development of new diagnostic tools of structure for dental rare diseases, such as amelogenesis imperfecta or dentinogenesis imperfecta.
Introduction
Dental development takes place between the sixth week of human embryonic development and the age of 18–25. 1 It is underpinned by epithelial–mesenchymal interactions between epithelial cells of the oral ectoderm and ectomesenchyme derived from the cephalic neural crest. It involves major developmental signaling pathways [Fibroblast Growth Factor (FGF), WNT signalling pathway (WNT), Sonic hedgehog (SHH), Nuclear factor kappa B (NF-KB), Transforming Growth Factor beta (TGFb)] and results in the establishment of highly specialized mineralized extracellular matrices (enamel, dentin, cementum, and alveolar bone), according to predefined patterns.2–4 In the early phase of tooth formation, dental epithelium and ectomesenchyme undergo segregation facilitated by a compact, in between, matrix layer, the basement membrane. This crucial separation endures until the advanced stage of dental bell formation. At that stage, specialized odontoblasts differentiate and secrete a predominantly type I collagen-based matrix known as predentin.
Dentinogenesis or dentin formation comprises a sophisticated interplay between several factors in the tissue, cellular as well as extracellular. It consists of odontoblast secretion of an extracellular matrix (ECM), the predentin, followed by mineralization. This matrix is composed of 90% of type I collagen and 10% of noncollagenous proteins (NCPs) and lipids. These NCP components are represented by phosphoproteins, proteoglycans, growth factors, enamel proteins, phosphatases (alkaline phosphatase), and proteases (matrix metalloproteinases and their inhibitors: matrix metallopeptidases [MMPs] and The tissue inhibitors of metalloproteinases (TIMPs)) that interact and build the scaffold that will support and initiate mineralization. 5 Following odontoblast differentiation, the basement membrane is enzymatically broken down by MMPs. 6 Consequently, the preameloblasts establish direct contact with both the predentin and the odontoblasts, which leads to their subsequent transformation into ameloblasts.
Ameloblasts play a crucial role in enamel formation by depositing various proteins into the enamel matrix. One of the primary proteins found in this matrix is amelogenin (AMEL), accounting for around 90% of the ECM composition. Additionally, nonamelogenin proteins such as enamelin (ENAM), tuftelin (TUFT), ameloblastin (AMBN), FAM83H, and proteases (such as MMPs) are also secreted by ameloblasts during the secretory phase. 7 Following the secretory phase, the ameloblasts enter a transition phase characterized by reduced secretion of matrix proteins and remodeling of their structure. Subsequently, the maturation phase ensues, during which the enamel matrix undergoes remodeling through the degradation of matrix proteins, ion transportation, and complete mineralization of the enamel. This process ultimately leads to the formation of mature enamel, the outermost layer of the tooth that serves as a protective barrier against mechanical and chemical insults.
The process of mineralization traps any defect during the stages of odontogenesis and allows the direct visualization and clinical examination, later in childhood or even in adulthood, of abnormalities in dental development such as abnormalities in tooth number, shape, size, structure, eruption, or resorption. These abnormalities exist most often in syndromes but may appear isolated because of the difficulty of associating them with other, sometimes discrete, clinical signs. Indeed, the borderline between an isolated or syndromic rare dental disease is very volatile.8,9
A targeted genetic study on rare diseases with oral manifestations, using high-throughput sequencing technologies (GenoDENT),9,10 developed by the Reference Center for Rare Oral and Dental Diseases (CRMR) of the University Hospitals of Strasbourg, currently exploring 567 genes, has allowed the identification (in a total cohort of 2232 patients including 878 index cases and 1354 related cases) of not only numerous pathogenic genetic variants (>60% of diagnostic rendering) but also numerous variants of uncertain significance (VUS or class 3 genetic variants—according to the American College of Medical Genetics 2019 classification).11–13 These VUS represent a major conundrum in terms of their significance and do not facilitate genotype/phenotype correlations. This complicates the establishment of a confirmed genetic diagnosis and further genetic counseling. 13 Thanks to the use of targeted sequencing or whole exome sequencing (WES), to provide a molecular and precision diagnosis in case of amelogenesis imperfecta (AI), dentinogenesis imperfecta (DI), or dentin dysplasia (DD) phenotypes, a positive diagnosis is reached in almost 60% of cases, less for DI/DD, but absent results and unsolved diagnostic wandering are still high (40%); however, in 10–20% of cases, VUS are identified.
Here, we present our research journey into finding in vitro solutions to unravel the pathogenicity of VUS during amelogenesis and dentinogenesis. We are studying the physiopathology of structural rare dental diseases (AI, DI, and DD). To do so, several research models have been described over the years, such as primary cells extracted from patients, cell lines, biopsies, genetically modified mouse models, extracted teeth, cell cocultures, and 3D cell cultures.14,15 In fact, in vitro culture models that allow to study epithelial–mesenchymal interactions are important for the understanding of dental development at the molecular level. 16 Two-dimensional (2D) monocellular monolayer culture cannot replicate the complex cell environment composed of dentin and enamel matrices or the processes observed during dentinogenesis (dentin formation) or amelogenesis (enamel formation). Thus, 2D cell culture techniques do not reproduce all the mechanical and biochemical signals present in vivo. 17 For example, during enamel maturation, in vivo, ameloblasts are positioned away from mesenchymal cells (odontoblasts), partially separated by the ECM and the proteins secreted by themselves and the mineralizing tissue. Subsequently, differentiated ameloblasts attach to the “hard” dentin surface and are potentially stimulated in many ways, including autocrine and paracrine pathways, cellular interactions, and cellular junctional communication modalities via mineral ion exchange. Subsequently, prior to terminal ameloblast differentiation, the basement membrane disappears. Recent studies have shown that collagen proteins, such as COLXVII, regulate ameloblast differentiation and are essential for the normal formation of Tomes’s processes, 18 or COLXVA1 has a modulator role in dentinogenesis and mineralization. 19 COLXVII deficiency disrupts epithelial–mesenchymal interactions, resulting in both defective ameloblast differentiation and enamel malformation. 20 However, no fully functional in vitro cell model is available so far, which would allow to study the molecular mechanisms by which the ECM mediates epithelial–mesenchymal interaction sites and thus modulates tooth formation in vitro. The limitations of 2D cell culture led to the use of animal models of gene inactivation that, although capable of mimicking the human pathophysiological situation, is also associated with ethical, technical, financial, and time constraints incompatible with a diagnostic report for a patient.17,21 3D models have been described to reproduce tissue physiology and phenotypes more accurately than 2D cultured cells. 17 Recently, some 3D “coculture” or single-cell population’ spheroids models have been developed to mimic this developmental process or the 3D tissue location of cells during dental development.14,15 These models use various cell populations, but authors have used primary cell or organ cultures and reconstituted ECM.14,15,21,22 The spherical structure of organoids with its radial symmetry offers opportunities for mapping cell functions in these gradients and 3D microenvironments. Most of the conventional microscopy methods and approaches commonly used for in vivo studies can also be used for imaging spheroids and organoids.14,17,23,24
To overcome these limitations, we propose innovative cellular organotypic models mimicking ameloblast–odontoblast compartmentalization during the bell stage of tooth development in a 3D cell environment, with a clear interface in between the two cell populations. In this study, our aim was to develop functional 3D cellular models made from mouse and human cells to recreate dental organogenesis. The resulting nonscaffolded 3D models allowed characterization at the molecular level of four different cell lines. We also demonstrated these models’ reliability compared with 2D cell monocultures. This model, which represents a low-cost, efficient, and reproducible cell culture system for cells responsible for the formation of the hard tissues of the tooth, will allow us to study VUS identified in patients with orodental rare diseases through inactivation by siRNA or induction of identified mutations by CRISPR/Cas9 technology. Through this experimental approach, we aim to gain insights into the functional consequences of these induced genetic variations in the context of dental development.
Materials and Methods
Patients, animal experiments, and ethics statements
This in vitro study was performed in correlation with our clinical study, which is registered at https://clinicaltrials.gov: NCT01746121/NCT02397824 and with the French Ministry of Higher Education and Research Bioethics Commission as a biological collection “Orodental Manifestations of Rare Diseases” DC-2012-1677/DC-2012-1002; it was acknowledged by the person protection committee. The oral phenotype of each patient is documented using the D[4]/phenodent registry protocol. 25
The animal experiments were performed in accordance with the French National and European Laws and Directives Concerning Laboratory Animal Housing, Welfare, and Experimentation and after approval from the CERBM-GIE: ICS/IGBMC Ethical Research Board. All animals were maintained and manipulated under animal protocols in agreement with the French Ministry of Agriculture guidelines for use of laboratory animals (C67-218-37-IGBMC « Mécanismes responsables de malformations osseuses et bucco dentaires: analyse de modèles murins » n° APAFIS#3957-2016020516359388v1).
Cell culture
Human dental pulp stem cells (hDPSCs) are multipotent stem cells harvested from soft living pulp tissue inside adult teeth. Immortalized hDPSCs were provided (courtesy of Dr. Papagerakis Petros, from the University of Saskatchewan, Canada, and Dr. Sylvie Babajko and Prof. Ariane Berdal, Paris, France; phenotypic characterization was performed as previously described). 26
The cells were grown in Alpha-Minimum Essential Medium (αMEM) medium + GlutaMAX-I with 1 g/L of
Mouse dental papilla cells (mDPCs) used were primarily obtained from molar tooth germs dissected from the mandibles of ED14.5 mice and immortalized as previously described,27,28 and they were provided by Dr. Malcom Snead (USC). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FCS and 1% PS at 37°C in a humid environment with 5% CO2.
Human ameloblast-like cells were initially characterized as ameloblastoma cells (AM-1) and were originally obtained from a 20-year-old female, immortalized with the HPV-16 vector, and clone selection was performed with 1 mg/mL G418, as previously described. 29 Cells were provided by Dr. Sylvie Babajko and Dr. Ariane Berdal (Paris, France) and were grown in keratinocyte Defined Keratinocyte-serum-free medium (KSFM medium) medium (ThermoFisher Scientific) complemented with 1 mg/mL G418 (Roche) for clone selection and 50 μg/mL gentamicin at 37°C in a humidified atmosphere with 5% CO2.
Mouse ameloblast-like cells, Ls8, an SV40-immortalized mouse ameloblast-like cell line, were primary isolated from the internal dental epithelium of the enamel organ of the mouse,30–32 and they were kindly provided by Dr. Malcolm L. Snead (USC). Cells were maintained in high-glucose DMEM (Gibco) containing 10% fetal bovine serum (Gibco) and 1% PS at 37°C in a humidified atmosphere with 5% CO2.
Odontoblast differentiation
hDPSCs and mDPCs were matured into odontoblast-like cells in a differentiation medium for 15–21 days. The differentiation medium was composed of the preceding mixture of 0.1 M dexamethasone, 5 mM glycerophosphate, 50 g/mL ascorbic acid, and 10 ng/mL TGF-β1, all from Sigma-Aldrich (Merck). The cells were incubated at 37°C in a 5% CO2 incubator. Cells were harvested using trypsin-Ethylenediaminetetraacetic Acid (EDTA) 0.25% and then used for different experiments.
Mouse tooth germs: Tissue preparation and histology
Few mouse embryos were obtained at E.17.5 of development, to analyze tooth germs at the bell stage when preodontoblast and subsequently preameloblast cells differentiate. Pregnant mice were euthanized with an intraperitoneal lethal injection of pentobarbital (100 mg/kg). Intracardiac perfusion with a fixative solution containing 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) pH 7.4 was then performed. Heads and maxillae were then dissected and postfixed by immersion in the same fixative solution overnight at 4°C. After extensive washing in PBS, the samples were dehydrated in increasing concentrations of ethanol and toluene and finally embedded in Optimal cutting temperature compound (OCT compound) (Scigen O.C.T. Compound Cryostat Embedding Medium, ThermoFisher) for frozen sections. Serial frontal sections of the mouse head were cut with a Cryostat™. Heads and maxillae frozen sections (E.17.5) and organoids harvested at day 21 were prepared for histology analysis by hematoxylin/eosin, Masson’s trichrome, and immunofluorescence. Briefly, organoids were washed with PBS after fixation and embedded in OCT for cutting (ThermoFisher). Then, sections of 30 and 50 μm were obtained using cryostat (Leica). Mouse sample slices were then treated with hematoxylin, eosin, and Masson’s trichrome staining kit (Merck).
Poly (d ,l -lactide-coglycolide) microscaffold synthesis
Microscaffolds were synthesized and kindly provided by Dr. Y. Artnz’s team (Inserm UMR_S 1121, Center de Recherche en Biomédecine de Strasbourg).
The porous poly (
D organoid formation using ultra-low attachment method without microscaffolds
Experimentations were made separately using cell lines from mouse (odontoblast-like cells, mDPC, and ameloblast-like cells, Ls8) and human (odontoblast-like cells, hDPSC, and ameloblast-like cells, AM-1) in the two-step culture method. Cells were detached from the cell culture flask with a trypsin treatment (trypsin 0.25% + EDTA 0.1%), resuspended into cell medium, and counted with a Neubauer chamber to be seeded at the concentrations mentioned below. First, odontoblast-like cells were cultured at a total cell number of 5 × 103 cells per well in an ultra-low attachment plate for 15 days (PrimeSurface® 96 V-shaped bottom, S-bio) with differentiation medium at 37°C in a 5% CO2 incubator (in 100 μL of medium). Then, 3 × 103 cells (ameloblast-like cells) per well were added with Keratinocyte Serum Free Medium (in 50 μL of medium) (Gibco, ThermoFisher) at 37°C in a 5% CO2 incubator (Fig. 1: same protocol, but without PLGA scaffolds). Organoids were observed under a light microscope and harvested at 15, 21, and 30 days.
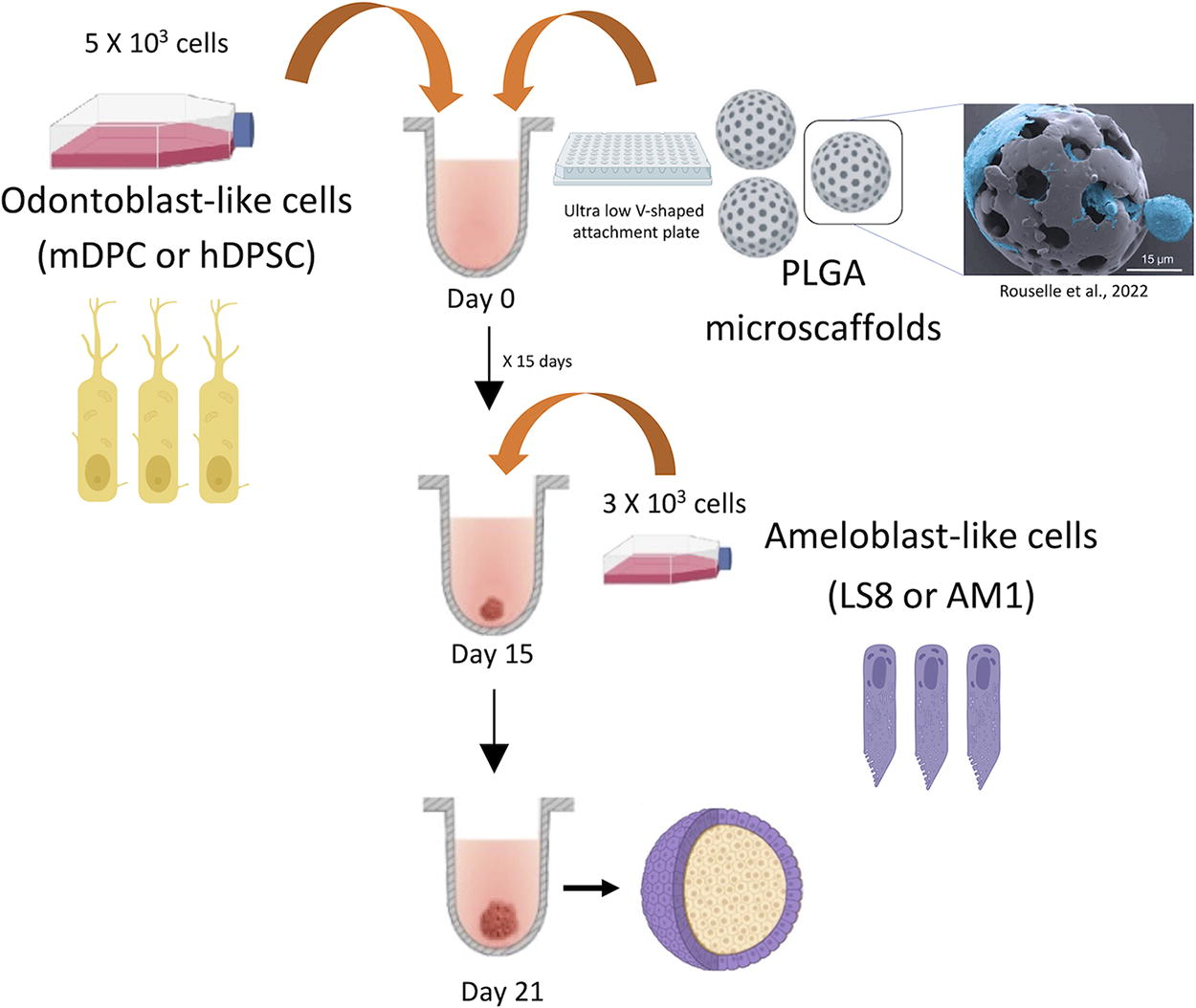
Schema and steps for organoids formation using ultra-low attachment plates and PLGA microscaffolds. Separate experiments were performed using cells from mouse (odontoblast-like cells differentiated from mDPC and murine ameloblast-like cells, Ls8) and from human (odontoblast-like cells differentiated from hDPSC and the ameloblast-like cell line, AM-1) in two main steps. After testing different organoid sizes, 5 × 103 mesenchymal cells and 3 × 103 epithelial cells were used for further experiments. Mesenchymal cells were first mixed with PLGA microscaffolds at a concentration of 10, 15, or 20 μg per well. The mix of cells and scaffolds was cultured in an ultra-low attachment plate for 15 days (PrimeSurface® 96 V-shaped bottom, S-bio) with differentiation medium at 37°C in a 5% CO2 incubator. The cultures were followed up under brightfield inverted microscope, and once the cells aggregated and started to differentiate (after 2 weeks of culture), 3 × 103 ameloblast-like cells/well without any scaffold were added with Keratinocyte Serum Free Medium (Gibco, ThermoFisher) at 37°C in a 5% CO2 incubator. The 3D cocultures were kept for one more week. Then, organoids were harvested (at 21 days of culture). hDPSC, human dental pulp stem cell, mDPC, mouse dental papilla cell, PLGA, poly (
Separate experiments were performed using cell from mouse (odontoblast-like cells differentiated from mDPC and murine ameloblast-like cells, Ls8) and from human (odontoblast-like cells differentiated from hDPSC and the ameloblast-like cell line, AM-1) in two main steps. After testing different organoid sizes, the following cell numbers were selected for further experiments: 5 × 103 mesenchymal cells (mDPC or hDPSC) and 3 × 103 epithelial cells (Ls8 or AM-1).
To form scaffolded organoids, 5 × 103 odontoblast-like cells/well were mixed with PLGA microscaffolds (at a concentration of 10, 15, or 20 μg per well). The mix of cells and scaffolds was cultured in an ultra-low attachment plate for 15 days (PrimeSurface 96 V-shaped bottom, S-bio) with differentiation medium at 37°C in a 5% CO2 incubator. The cultures were followed up under a brightfield inverted microscope, and once the cells aggregated and started to differentiate (after 2 weeks of culture), 3 × 103 ameloblast-like cells/well without any scaffold were added with Keratinocyte Serum Free Medium (Gibco, ThermoFisher) at 37°C in a 5% CO2 incubator. The 3D cocultures were maintained for 1 more week. Then, organoids were harvested (at 21 days of culture; Fig. 1).
Organoid cutting preparation
Organoid fixation was performed with 4% PFA. Briefly, medium was taken out, leaving formed organoids on the bottom of plates, and then they were washed twice with PBS and incubated with PFA for 30 min at room temperature (RT). Then, the PFA was discarded, and wells were washed with PBS twice. With the help of a 200 µL tip, organoids were isolated and stored in a 2 mL tube. Organoids were incorporated into a block of OCT and placed at −20°C to freeze. The block was then cut with a cryostat, and 500 µm sections were set on glass for confocal microscopy.
Immunohistochemistry and immunofluorescence
Immunohistochemistry was performed on 2D cell cultures, organoid sections (50–100 µm), and mouse tissues harvested from mouse embryos (mouse tooth germs at bell stage). All sections (cells, mouse tissues, and organoids) were fixed with PFA 4% and mounted on glass slides. Following a 15-min PBS-Tween 0.2% treatment, cells were saturated with Bovine serum albumin (BSA) 0.5% and Triton X-100 0.1% in PBS (PBS-BT) for 15 min at RT. Primary antibodies (Supplementary Table S1) were diluted to 1/100 in PBS-BT and incubated for 1 h at RT. Secondary antibodies (Supplementary Table S1) were diluted to 1/250 or 1/500 in PBS and then incubated at RT for 1 h. Cells and tissues were washed thrice with PBS. Then, organoids slides or cells were incubated in the phalloidin solution for 15 min at RT. Phalloidin was prepared by mixing 30 µL of dimethyl sulfoxide (DMSO) with the vial powder and diluting it to 1/1000 in PBS-1% BSA. After washing the slides twice for 5 min at RT, one drop of 4′,6-diamidino-2-phenylindole (DAPI) solution was added to each slide. Finally, a cover glass was deposited on the top of the slides after washing once more with PBS and adding one drop of mounting medium. Mounted slides were observed with an epi-fluorescence microscope (Leica DM4000B).
Organoid clearing
Organoid sections (100 µm) or full-size organoids (400–600 µm) were transferred from the postfixation solution (4% PFA) directly into the glass vial, and the first clearing solution was immediately added [the first clearing solution is 50% (vol/vol) tetrahydrofuran] using a Pasteur pipette. Then, the glass vial was placed onto a turning wheel. Samples were covered with aluminum foil to keep them in the dark, and all the clearing steps were performed in a fume hood at RT, being rotated using the wheel at constant speed (20–40 rpm). The clearing solution is then discarded, and the next clearing solution is immediately added (DCM). A previously described protocol 33 was further used for all clearing solutions, until the last clearing solution (LCS) was added. To achieve good transparency, LCS incubation time was optimized according to the thickness and type of tissues, 33 showing that 15–20 min Organic solvent dibenzyl ether (DBE) incubation was sufficient. Finally, imaging was performed using confocal high-speed multispectral spinning disk microscopy.
Confocal high-speed multispectral spinning disk microscopy
The preferential system used to image our samples was the confocal high-speed multispectral spinning disk microscopy of immunofluorescent cells and organoids. We used the multispectral spinning disk confocal microscope on the inverted Leica DMI8 for brightfield and fluorescence (not DIC and Ph contrast, spinning head: CSU W1, XY mot stage for multilocation recordings, Z piezo stage for fast z stack [range 300 m], camera: Evolve 512: 512 × 512 pixels; pixels size: 16 m, and Orca Flash 4.0: 2048 × 2018 pixels; pixels size: 6.5 m) and used a 63 × 1.4-HCX PL APO Lambda blue or 100× HC PL APO 1.47-NA objective lens. The original grayscale photos from each fluorescence channel were converted to color; contrast, brightness adjustments, and unsharp masking filters were used to create the figures (Figs. 2–4, 8).
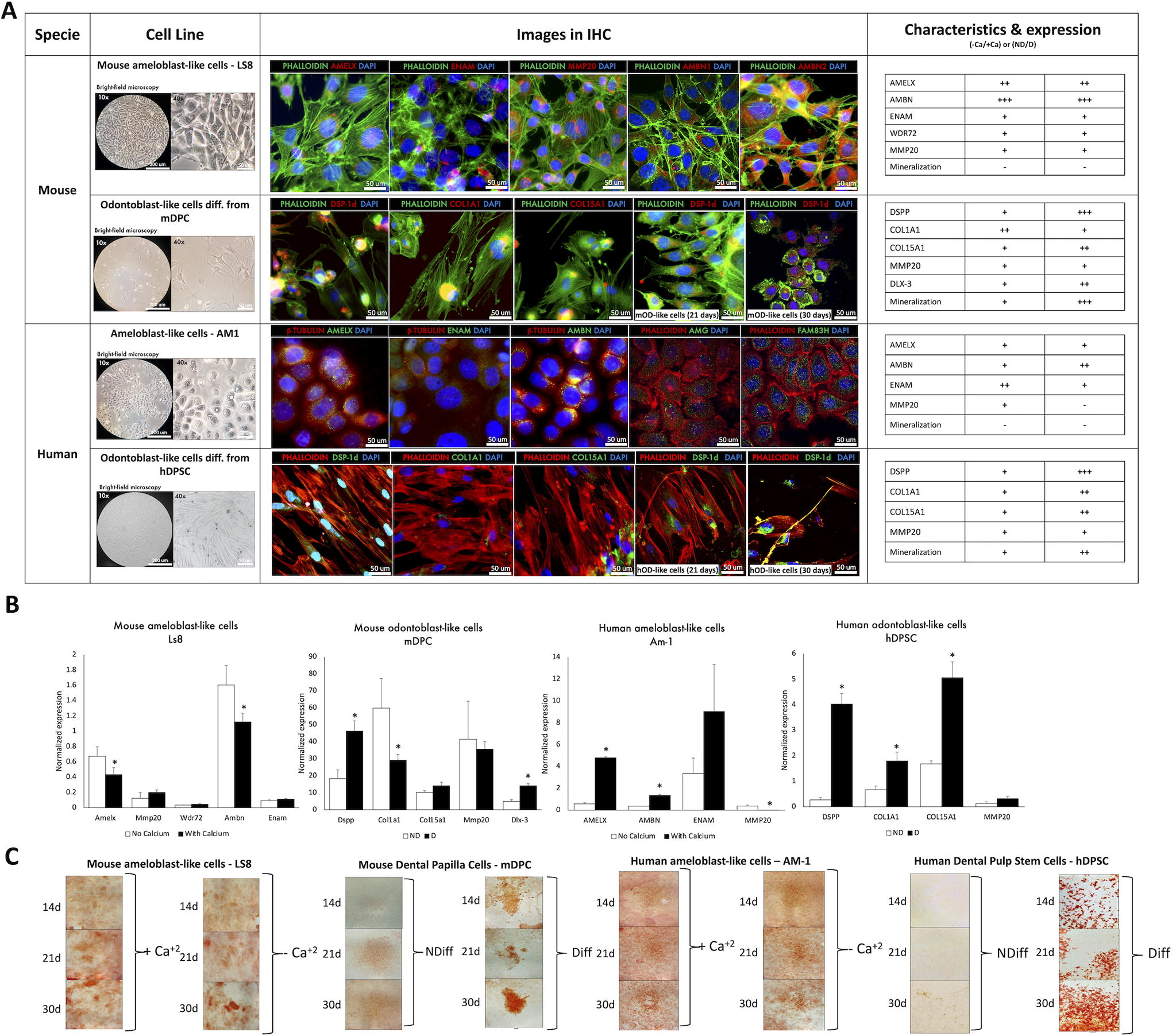
Odontoblast-like cells differentiated from dental pulp stem cells and ameloblast-like cells: cell morphology, mineralization, and mRNA and protein expression.
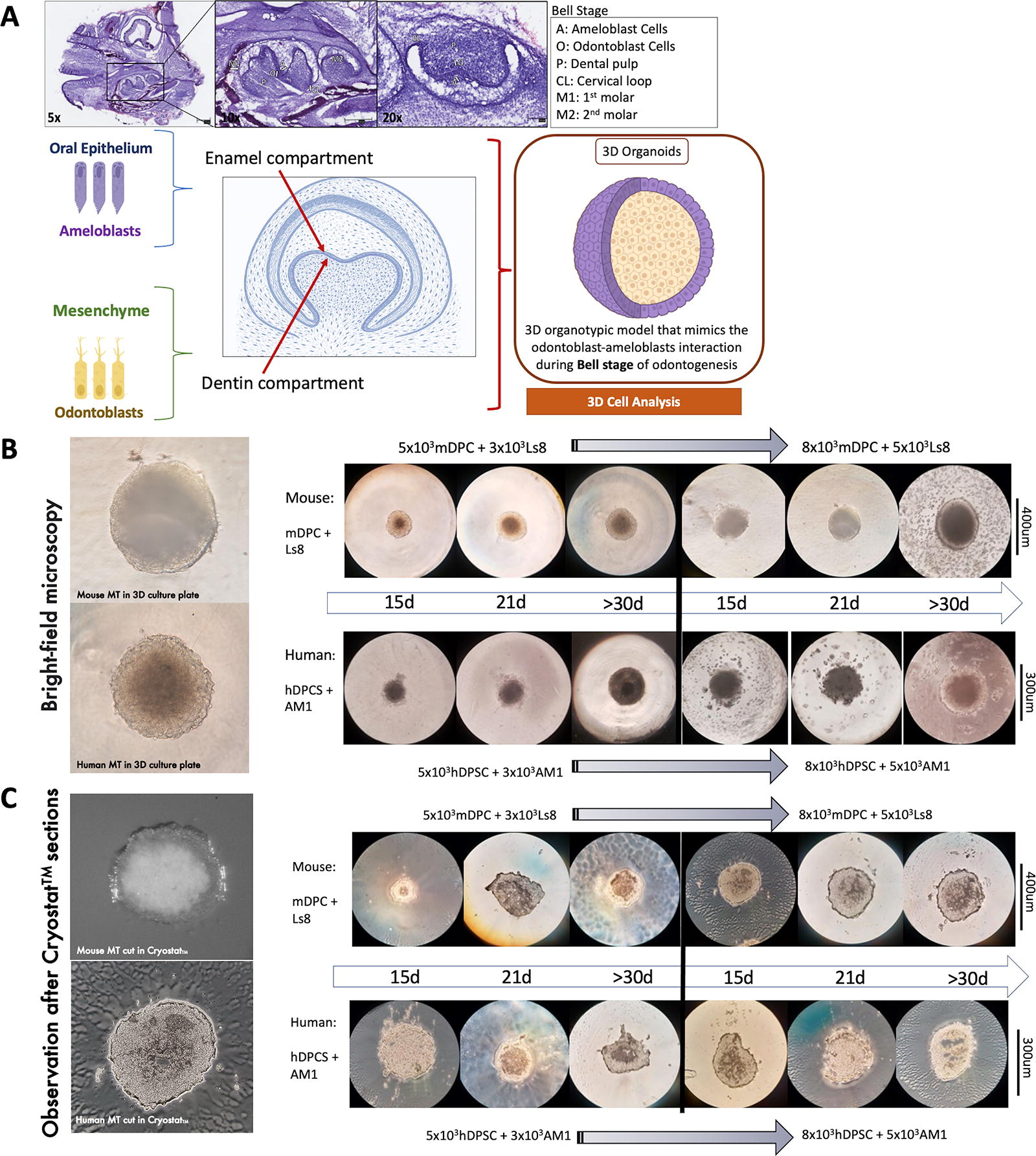
3D organotypic cellular models of mice and human cells show a bilayer distribution of epithelium and mesenchymal cells.
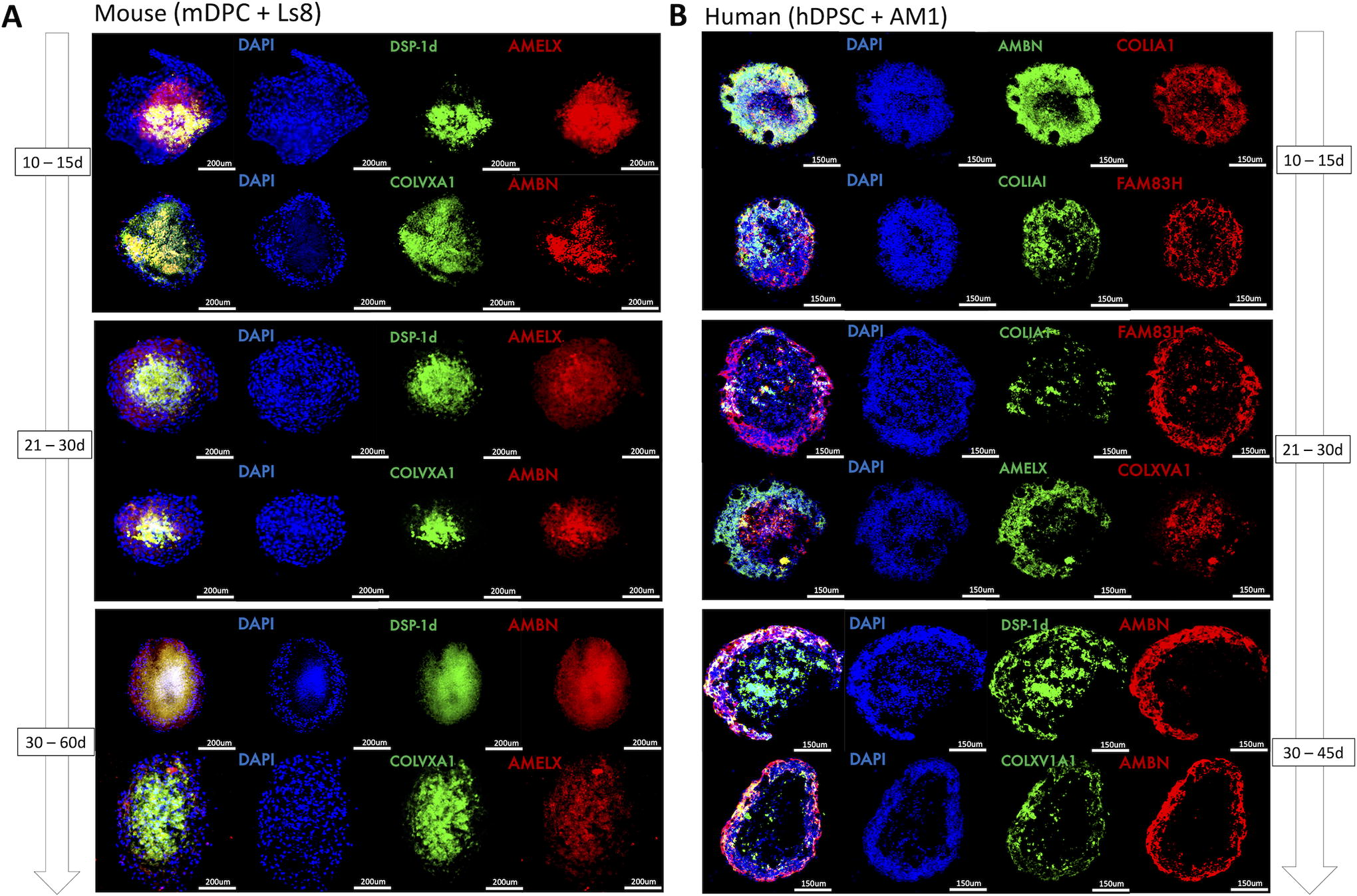
3D organotypic cellular models recreate dentinogenesis and amelogenesis molecular patterns of expression found in odontogenesis.
RNA extraction
TRIzolTM Reagent (ThermoFisher) was used to extract total RNA from cells and organoids until the separation phase steps, and then the RNeasy® Plus Micro kit was employed (Qiagen) in the way to keep DNA and proteins from the same samples. First, 400 µL of TRIzolTM was added to the 24-well culture plate after the media had been removed, and the cells were scratched with a point. The leftover plate cells were saved from the lysate by being placed in 100 µL of PBS, which was then mixed. The lysate was treated with 0.1 mL of chloroform for 3 min before being centrifuged at 12,000 g for 15 min at 4°C. The aqueous phase containing the RNA was separated from the other two phases and placed in a spin column with gDNA Eliminator. The manufacturer’s recommendations were followed for the next steps (RNeasy Plus Kits, Cat. No./ID:74034, Qiagen). Briefly, tgDNA Eliminator column was centrifuged at a speed of 8,000 g for 30 s. The flow-through was combined with one volume of 70% ethanol, transferred to the RNeasy spin column inserted in a 2 mL collection tube, and centrifuged for 15 s at over 8,000 g. After removing the flow-through, 700 µL of Washing buffer 1 (BW1) buffer was added to the column. This centrifugation process was repeated using 500 µL of mild washing buffer RPE (RPE buffer) buffer and then 500 µL of 80% ethanol. The column was put into a clean collecting tube and centrifuged for 5 min at 12,000 g. A fresh collecting tube was used to hold the column, and 14 µL of RNase-free water was added right to the spin membrane’s center. To elute the RNA, one final minute of full-speed centrifugation was performed. The NanoDrop 1000 was used to measure the total RNA concentration (ThermoFisher). The concentration was then adjusted to 200 ng/µL for all samples.
Reverse transcription PCR
Using the SuperScriptTM II RT (ThermoFisher) according to the manufacturer’s recommendations, the mRNA was reverse transcribed. Two mixtures were made: one contained 250 ng of a random primer (1 µL), 1 µg of RNA diluted in 9 µL of sterile DW, and 2 µL of dNTP (5 mM); the other contained 4 µL of 5× first-strand buffer, 0.1 µL of DTT, 1 µL of RNaseOUT, and 1 µL of SuperScript. Mix 1 was heated to 65°C for 5 min, chilled on ice before being added to mix 2, and then incubated for 50 min at 42°C before the final incubation of 15 min at 70°C. At the end, 40 µL of DNase-free water was added.
Quantitative PCR (cDNA)
The cDNA samples were subjected to qPCR to measure mRNA expression. RealPlex 2 qPCR Real-Time PCR ThermoCycler was used to amplify and analyze PCR (Eppendorf). Using the LightCycler® 480 SYBR Green I Master, amplification reactions were carried out (Roche Life Science). In all samples, GAPDH served as the endogenous RNA control (housekeeping gene). Primer sequences for the murine genes were bought from the manufacturer (Merck). After the expression of housekeeping genes was normalized, the relative expression level was determined. All primer sequences (for human and mouse) are presented in Supplementary Table S1.
Alizarin Red assay
Alizarin Red solution was used to specifically stain calcium deposits in a vivid orange-red color (Reference #A5533, Merck). In order to make the staining solution, 2 g of Alizarin Red was dissolved in 100 mL of DW. The pH was adjusted using 0.1% ammonium hydroxide, reaching 4.1–4.3. The 24-well plate’s medium was removed, and PBS was used to wash the cells. The cells or organoid sections were covered with PFA (4%) for 30 min. Cells were washed with DW only after PFA was removed. After adding the staining solution, 200 µL per well, the plate was incubated at RT in the dark for 45 min. Before adding PBS, Alizarin Red was gently removed, and the cells and organoid sections were washed thrice with DW.
Scanning electron microscopy
Organoids were mounted on aluminum scanning electron microscopy (SEM) stubs and sputter-coated with a gold–palladium alloy (20/80 weight %) using a Hummer JR sputtering device (Technics) for 7–15 min after being dehydrated in a series of alcohol solutions (50%, 70%, 90%, and 100%) (Technics). The morphological characteristics were determined using a Quanta 250 FEG scanning electron microscope (FEI Com-190 pany) at 10 kV acceleration voltage of the electrons. Energy dispersive X-ray analysis (n = 5) was performed with a working distance of 10 mm and an acquisition time of 60 s.
Transmission electron microscopy
The samples were fixed by immersion in 2.5% glutaraldehyde and 2.5% PFA in cacodylate buffer (0.1 M, pH 7.4) and then washed in cacodylate buffer for further 30 min. The samples were postfixed in 1% osmium tetroxide in 0.1 M cacodylate buffer for 1 h at 4°C, rinsed with cacodylate buffer 0.1 M (2 × 10 min) and with water (2 × 10 min), and then immersed in uranyl acetate at 4% for 2 h at 4°C. Samples were dehydrated through graded alcohol (50, 70, 90, and 100%) and propylene oxide for 30 min each, before being embedded in Epon 812. Semithin sections were cut at 2 µm, and ultrathin sections were cut at 70 nm (Leica Ultracut UCT), contrasted with uranyl acetate and lead citrate, and examined at 70 kv with a Morgagni 268D electron microscope (FEI Electron Optics). Images were captured digitally by a Mega View III camera (Soft Imaging System).
Statistical analysis
Statistical analysis was performed using the pairwise analysis of variance test and post hoc Tukey’s test. Statistical significance level was considered for p < 0.05. Data were analyzed using PRISM 6.0 (GraphPad). All experiments have been performed at least in biological and experimental triplicates.
Results
Characterization of mouse and human cell lines morphology and molecular signatures: Amelogenesis and odontogenesis-related gene and protein expression
To validate mouse (mDPC and Ls8) and human (hDPSC and AM-1) cell lines as suitable cellular models for in vitro assessment of odontoblasts and ameloblasts, we investigated mRNA and protein expression of the main molecules implicated in dentinogenesis and amelogenesis. Both mouse and human cell lines exhibited an adequate profile of protein expression of the molecules of interest, which are involved in these two main processes during odontogenesis. According to the analysis of immunofluorescence and mRNA expression in the four cell lines (Fig. 2, Supplementary Fig. S1), mDPC and hDPSC expressed dentinogenesis-associated proteins such as dentin sialoprotein (DSP), COLIA1, and COLXVA1 (Fig. 2A, Supplementary Fig. S1B, D and their expression was increased after differentiation. Ls8 and AM-1 expressed amelogenesis-associated proteins such as AMELX, ENAM, AMBN, and MMP20 (Fig. 2B, Supplementary Fig. S1A, C), and this was according to previous data.34–36 COLIA1 and COLXVA1 19 were highly expressed in the ECM of dental pulp cells. Morphology and growth behaviors of Ls8 and AM-1 were compatible with an epithelial cell profile,37,38 while for mDPC and hDPSC, patterns of expression and morphological characteristics were closer to mesenchymal pulp cells described previously in the literature34–36 (Fig. 2A, B). Before differentiation, hDPSC showed a flatter and spindle shape close to a typical ectomesenchymal morphology, whereas cells after differentiation induction in vitro [human Odontoblast-like cells (hOD)-like cells and mouse Odontoblast-like cells (mOD)-like cells] were more compact with some visible features in their cytoplasm and protein expression, shown in the immunostaining images, compatible with 2D cell odontogenic differentiation. This differentiation feature of the mesenchymal cells was also observed in mDPC but was more modest (Fig. 2A, Supplementary Fig. S1B). Notably, both epithelial cell lines (Ls8 and AM1) demonstrated good proliferation potential, as evidenced by their rapid growth from an initial confluence of 30% to ∼90% within a span of 10 days. Odontoblast-like cells, especially human, demonstrated limited proliferation capacity after differentiation, which suggests that the differentiation of hDPSC into hOD-like cells was successful. The differentiation was further characterized by mRNA expression profiles and mineralization (Fig. 2B, C). Overall, dentinogenesis- and amelogenesis-associated mRNA expression was more pronounced in human cell lines, with significantly increased expression of DSPP, COL1A1, and COL15A1 for dentinogenesis and AMELX in AMBN for amelogenesis (Fig. 2B). Very low expression of MMP20 was detected in odontoblast cell lines as expected and high expression in ameloblast-like cells. The mineralization was efficient for both mouse and human cell lines, whereas no significant difference was observed for ameloblast-like cells (AM-1 and LS8), as expected (Fig. 2C). The results are summarized in Figure 2A.
Molecular expression patterns of amelogenesis and dentinogenesis processes have been previously characterized in mouse and human tooth development.34–36 Key protein expression of dentinogenesis (DSPP, COLIA1, and recently described COLXVA1) 19 and of amelogenesis (AMELX, ENAM, MMP20, and AMBN) was evaluated in organoids after 15, 21, and 30 days of 3D culture (Fig. 3, 4). In the early bell stage, odontoblasts can be observed in the subjacent area of preameloblasts, interacting mostly via the basement membrane and before the secreting matrix. In the late bell stage, the absence of the basement membrane is correlated with the differentiation of odontoblasts, the presence of predentin, and the consequent changes in morphology and protein expression of enamel proteins by preameloblasts. Scaffold-free 3D organotypic cultures were constructed in two main steps and then harvested at six time points (three early differentiation timeslots and three late differentiation timeslots) (Fig. 3). The first step was conducted with mesenchymal cells only (mDPC for mouse organoids and hDPSC for human organoids). Then, in the second step, ameloblast-like cells (Ls8 for mouse and AM-1 for human) were added to form two-layer organoids, mimicking the two-cell compartment interaction during the bell stage (Fig. 1, 3).
After microscopy observation of the resulting structures (Fig. 3B, C), molecular expression patterning of cell organoids and of the two cell populations and their interface was evaluated by immunofluorescence and confocal microscopy (Fig. 4, 5). The initial analysis by IHC showed expression of dentinogenesis-associated proteins (DSP-1 domain of DSPP, COLIA1, and COLXVA1) by odontoblast-like cells mainly in the core of these 3D structures, and amelogenesis proteins (AMELX, AMBN, ENAM, MMP20, and FAM83H) on the periphery, expressed by ameloblast-like cells (Fig. 4C). The two layers of human organoids composed of odontoblast-like cells and ameloblast-like cells were clearly distinct, whereas, in the mouse organoids, this distinction was more modest, and some overlap of the markers in this interface was observed. These results confirmed that differentiation medium allowed mDPC and hDPSC to mature into odontoblast-like cells with characteristic protein expression profiles after 21 days. Ameloblast–odontoblast interface and populations were analyzed, and we were able to differentiate both of them in mouse and human organoids, though specific molecular markers are associated with enamel or dentin matrix, before mineralization (Figs. 4A and B). Some specificities of the two cell compartments and the differentiation and mineralization were next evaluated in 3D by confocal and epifluorescence microscopies and showed a clear molecular expression, depending on the cell distribution (Fig. 4, 5). An increased differentiation of the odontoblastic core was also observed when both cell populations were cocultured14,39 (supported by unpublished data), both in human and mouse organoids (Fig. 5).
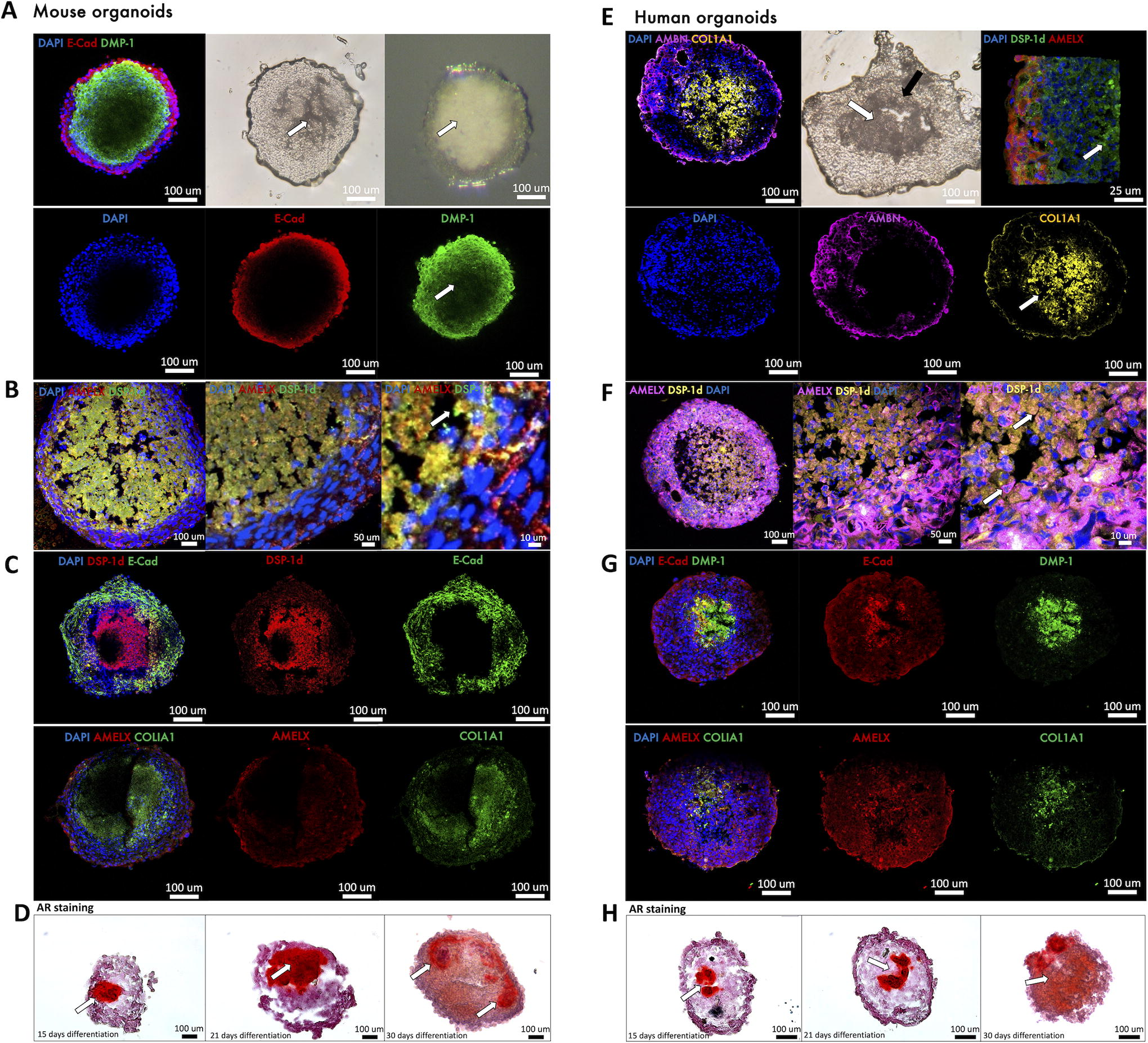
Evaluation of the ameloblast-like cells and odontoblast-like cell compartments in mouse and human organoids. Confocal microscopy was performed on full mouse organoids or thick sections (150–200 μm) that were first stained and then cleared, in order to complement the thin cryostat sections shown in Figure 4.
Thick sections (150–200 µm) or full organoids were stained and then tissue clearing was performed, and they were observed under confocal microscopy (Fig. 5), in order to complement the compartmentalization of the organoids and the distinction between the two cell populations. A specific structural marker to epithelium, E-cadherin, and AMELX or AMBN was observed on the outer layer of the organoids (Fig. 5A, B, C, E, F, G). This correlated with strong cell-to-cell adhesion in the epithelial compartment and to enamel matrix secretion, but no mineralization (Fig. 5D, H), both in murine and human organoids (Fig. 5). In mammals, matrix proteins other than collagen include DSP, dentin phosphoprotein (DPP), and dentin matrix protein 1 (DMP1). In particular, DSP-1 domain and DPP, both of which are produced from a common gene, DSPP, are recognized as dentin-specific matrix proteins. DMP1, DSP, and the DPP are the noncollagenous components that regulate dentinogenesis. DMP1 expression was localised at sites of mineralization and it was strongly expressed by the odontoblastic core of both murine and human organoids (Fig. 5A, G).
Outside structure and cell organization of organotypic cell cultures by SEM showed ameloblast-like cells on the surface with differential behavior between human and mouse models
Murine and human organoids without microscaffolds, conceived by the ultra-low attachment method, showed distinct surface characteristics on SEM analysis (Fig. 6). All of these organoids, whether human or mouse, were analyzed from 21 to 30 days of culture, which was sufficient time to observe a differentiation of the odontoblastic cells in the core, the pulp cells into odontoblast-like cells, and sufficient condensation and interaction with the surface epithelial compartment cells (Fig. 6A, B). SEM images show that human organoids (Fig. 6A) are smaller and less compact than murine organoids (Fig. 6B). Outer epithelial cells present a suitable morphology with ameloblast-like cells, with microvilli on the surface and strong interactions between them, but in certain areas, an exposure of the odontoblast layer and an onset of spheroid degradation (Fig. 6B) are observed. This might be correlated with the manipulation of the spheroid’s structures in all the processes of fixation, staining, and/or observation. In the case of mouse cell organoids, it is important to note a higher cell density; they are more compact with some collagen fibers possible to observe under the epithelial layer (white arrows), and their structure is more uniform than human organoids (Fig. 6A, B). In both types of organoids, the surface is almost completely covered by epithelial cells (yellow arrows). In human organoids, more cellular debris and apoptotic cells are observed, even on the surface, compared with mouse organoids, which is confirmed by transmission electron microscopy (TEM) analysis images (Fig. 7).
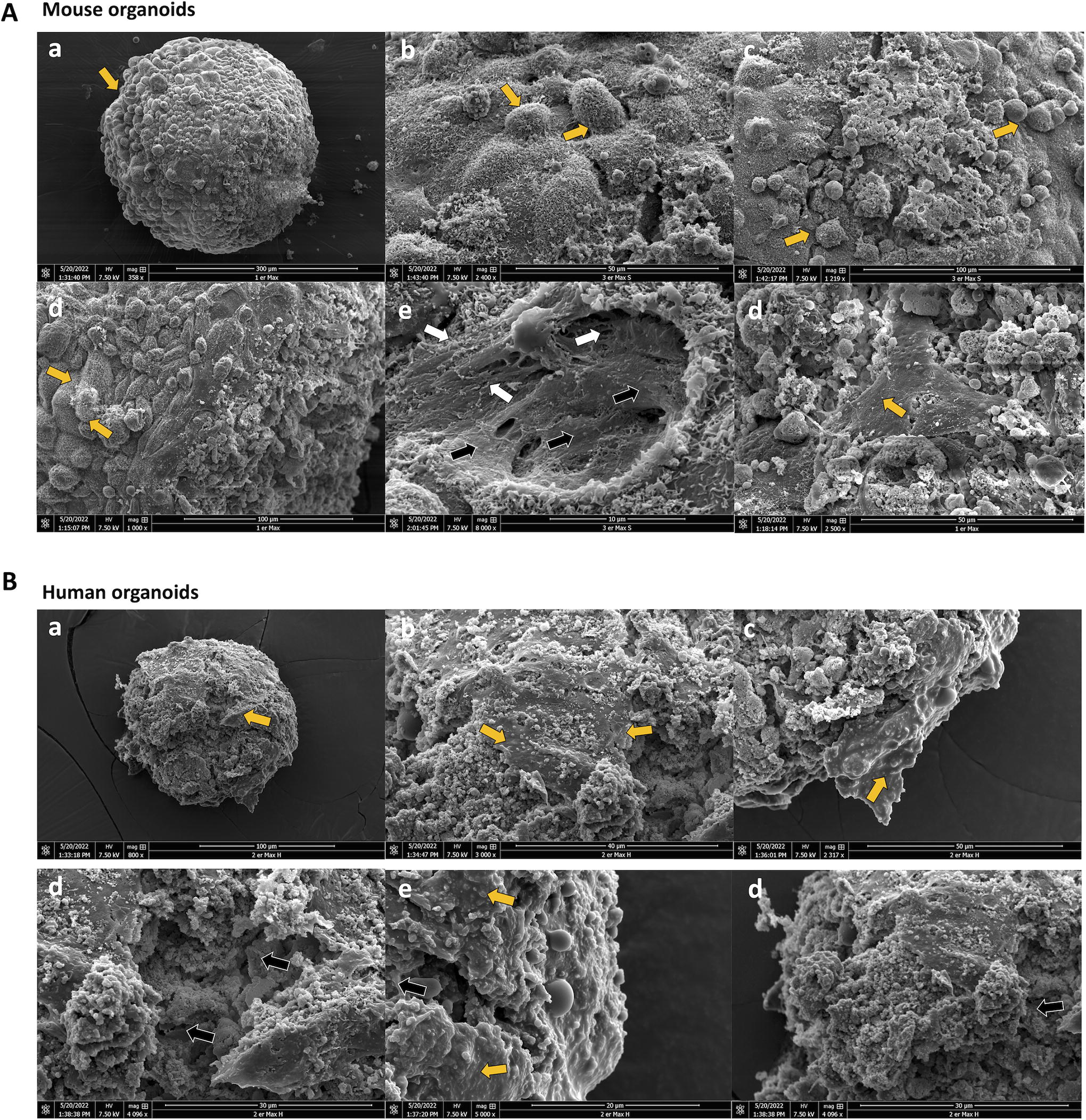
SEM evaluation of the outside structure and cell organization of organotypic 3D cell cultures.
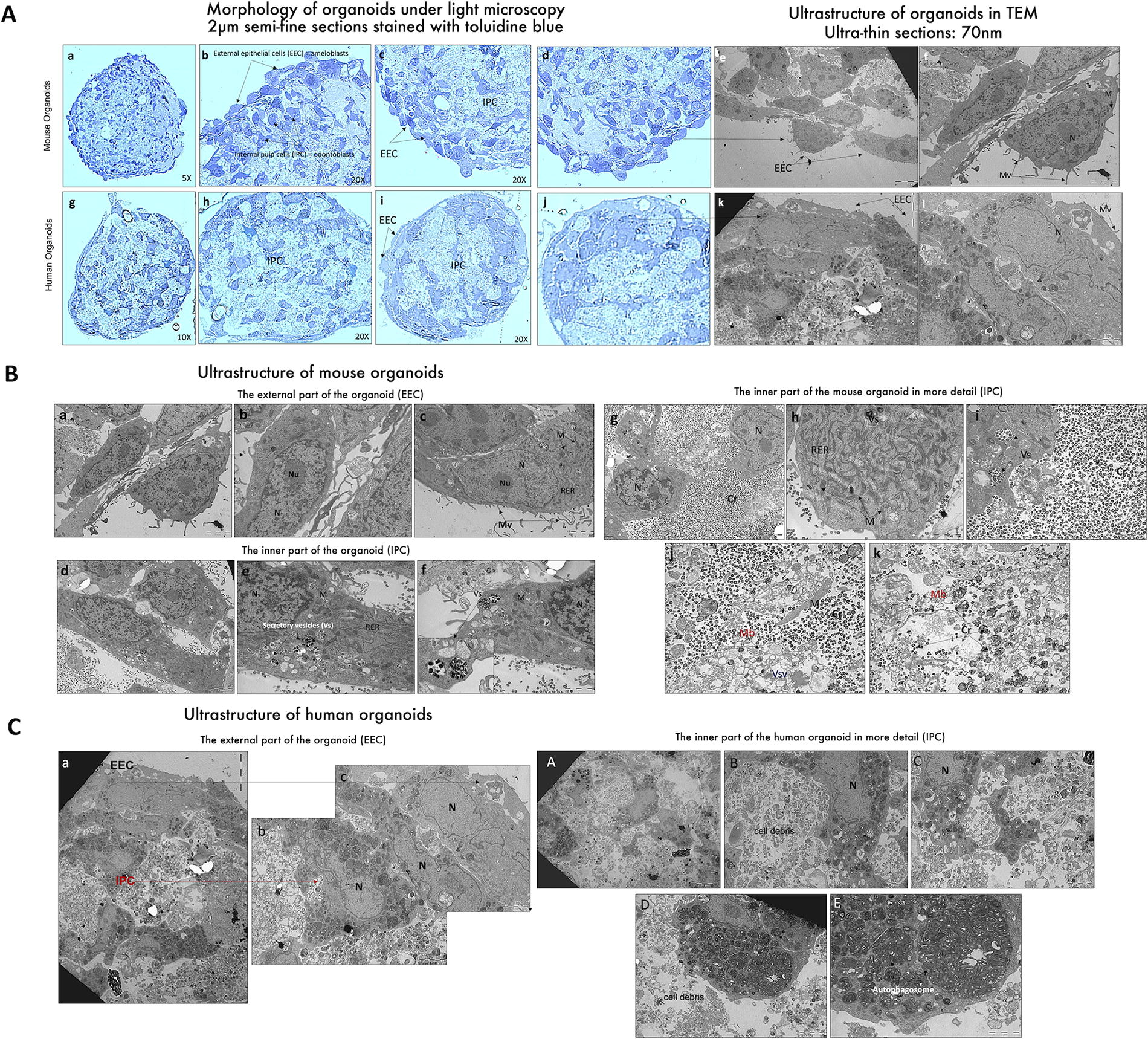
Outside and inside ultrastructure of organotypic 3D cell cultures.
Inside ultra-structure of organotypic 3D cell cultures assessed by TEM and SEM showed cell-to-cell interactions of ameloblast-like cells and odontoblasts
Before Transmission electron microscopy (TEM) analysis, in order to study the ultrastructure on both cell populations, we analyzed, on 2 µm histological sections, the inner cell organization of the two organoid types. Two main layers of cells could be observed: an outer layer showing external epithelial cells (EECs, ameloblast-like cells), surrounding the entire surface of the organoid, and an inner layer showing internal mesenchymal-pulp cells (IPC, odontoblast-like cells) (Fig. 8A). Then, TEM analysis of mouse organoids showed that the EEC (Fig. 8B) consists of two layers of well-differentiated epithelial cells (elongated appearance of the cell with a nucleus [N] and mitochondria [M] and the presence of cellular membrane protrusions that can be described as microvilli [Mv]), visible on the outer surface. More detailed analysis demonstrated that, in mouse organoids (Fig. 7B, C), the ultrastructure of EECs (ameloblast-like cells) at high magnification shows cells rich in rough endoplasmic reticulum (RER) and mitochondria (M) and a considerable number of cytoplasmic extensions (Mv) and nuclei (N) with nucleoli (Nu). Instead, in human organoids (Fig. 7C), cells in the external epithelial layer do not present similar characteristics of cell differentiation, as murine organoids and some lipid-filled cells and electron-dense vesicles are observed consistent with cellular stress. This structural instability of human organoids appears to be related to the odontoblastic core fragility and the lack of interaction between the two cell compartments, which show to be more specific and stronger in murine organoids. At the inner part of mouse organoids (Fig. 7), odontoblast-like cells are releasing vesicle contents (crystals) by exocytosis into the external environment to form the predentin framework. The richness of the cell cytoplasm in RER and mitochondria (M) was noted. mDPC, forming the core of organoids, differentiated into secretory cells, capable of releasing a collagen-rich matrix and vesicles loaded with crystal particles. Further analysis might be conducted to study the type of mineralized particles present in these vesicles. These odontoblast-like cells are rich in RER and Golgi apparatus, reflecting a high capacity for exocytosis, and subsequently produce a very dense predentin framework from electron-dense intracytoplasmic secretory vesicles (Vs). A few collagen fibers scattered throughout the predentin framework were also visible. At high magnification, the predentin like matrix is clearly visible, consisting of crystal particles (Cr) and a few degenerating inner pulp cells (ghost mitochondria [M], cytoplasmic membranes [Mb], and empty secretory vesicles [Vsv]). Considering the ultrastructure and the internal structure of the inner and outer epithelial and pulp cells of human organoids (Fig. 7), first we observed a halt in cell differentiation in the outer epithelium. The cells are in distress, filling up with small vesicles (lysosomes) and large vesicles, a sign of cell degradation. The inner part of organoids clearly shows cell degradation, with the center filled with cell debris instead of predentin or dentin (collagen fibers and/or crystal particles; Fig. 7B, C). We observed these large electron-dense vacuoles (autophagosomes) at high magnification mainly in the core of human organoids, suggesting a very high cell stress and cell death of hDPSC.
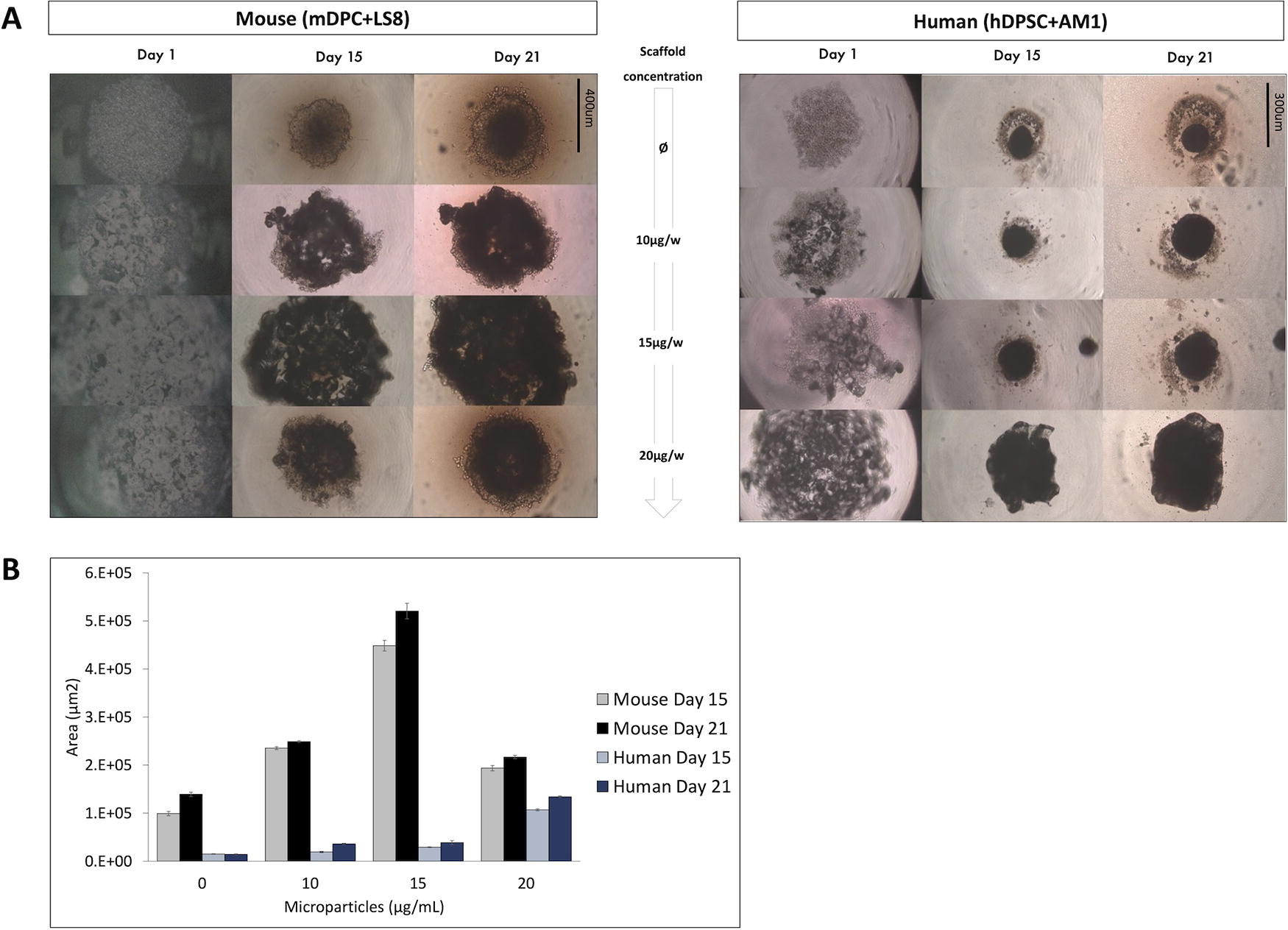
Organoid size increases in proportion to the amount of initial PLGA microscaffolds added to the odontoblastic core.
Microscaffolds enhanced organoid size and structural stability but not the cell distribution for human organoids, which still showed the expression of dentinogenesis and amelogenesis molecules
To improve the stability and cell viability of human organoids, in particular the core of odontoblast-like cells, PLGA microscaffolds were used to support the cell growth (Fig. 8, 9). It has been recently shown that these scaffolds allowed to improve cell survival and enhance the 3D culture of hDPSC 39 in organotypic cultures. Three different concentrations of PLGA microscaffolds were tested: 10, 15, and 20 μg per well. First, light microscopy was used to study cell organization in 3D (Fig. 8A), and organoid size was quantified using ImageJ (Fig. 8B). Interestingly, at the same cell numbers, human organoids were smaller than mouse organoids when they were scaffolds free, but their size increased when 20 μg per well of microparticles was added. However, in mouse organoids, lower cell aggregation compared with scaffold-free organoids was observed (Fig. 8A).
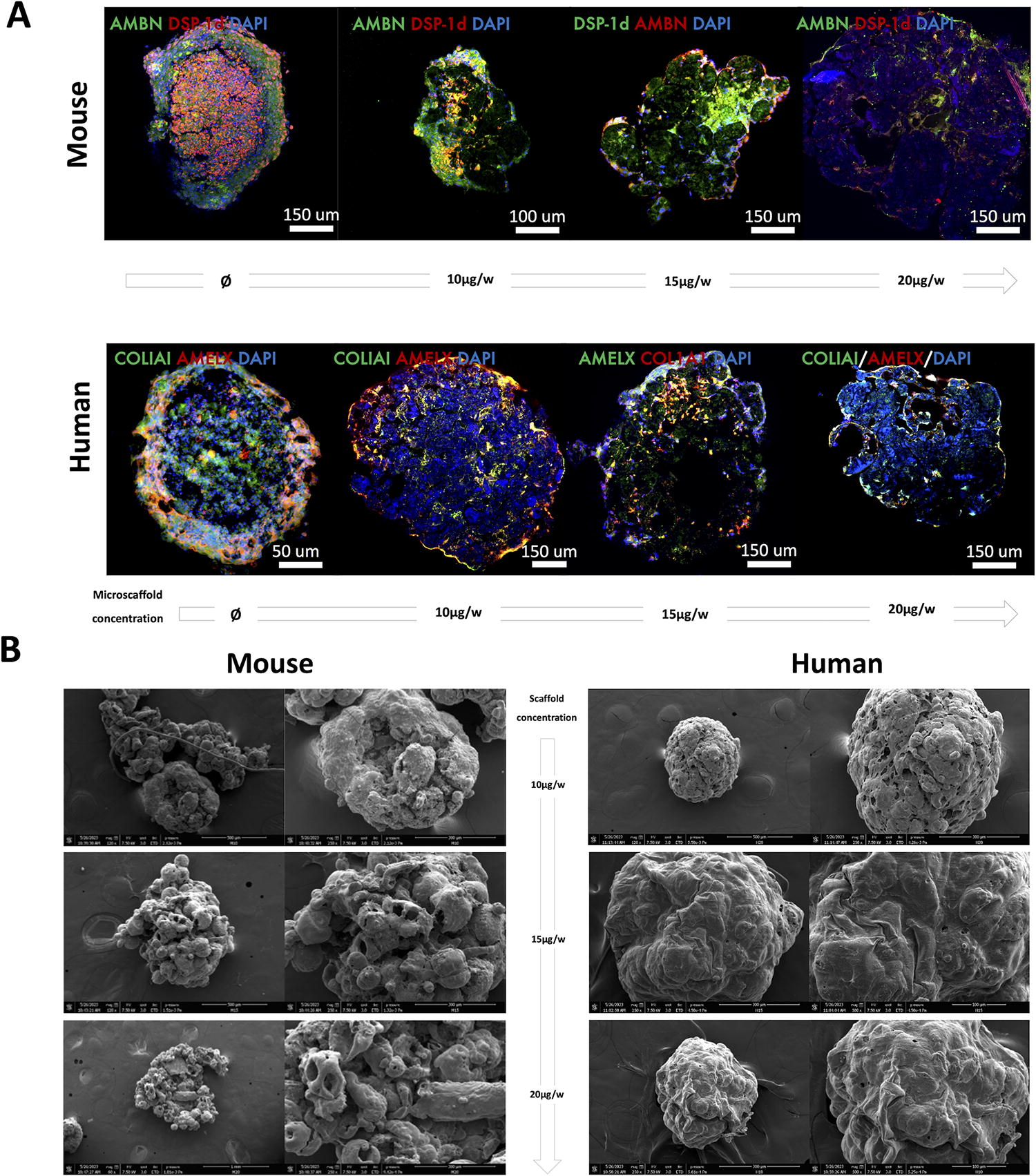
Microscaffolded organoids keep enamel and dentin protein expression, but PLGA scaffolds alter its distribution.
Even though the structure of microscaffolded organoids appeared more stable in time for human organoids, immunofluorescence analysis suggests that the organization and distribution are less distinct between the two cell populations (Fig. 9A). However, protein expression and cell morphology confirmed cell viability of cells in the core and on the periphery, which was importantly decrease in human organoids without scaffolds. Finally, according to MEB analysis (Fig. 9B), spaces in between each round scaffold allowed the penetration of ameloblast-like cells in the core of the organoids. Concentration of microscaffolds had a differential effect on the distribution of cell population, which was not linear. The shape of organoids seems less homogeneous and contains microporosity induced by interaction between PLGA microscaffolds and cells (Fig. 9B), which is especially pronounced for mouse microscaffolded organoids.
When considering the advantages that the presence of microscaffolds can bring to both mouse and human organoids, it is important to note that manipulation of these 3D cellular structures is easier when using these scaffolds. Even when using the same 3D culture technique and in two stages, the two cell populations tend to mix much more when using microscaffolds (Fig. 8A, 9A). Dentin protein expression remains mainly in the center of these organoids, and enamel protein expression remains more peripheral but with less distinction, as compared with scaffold-free organoids (Fig. 9A). Mouse organoids already had very good organization and protein expression without scaffolds, so the contribution of these microstructures does not seem to add value to this type of organoids. In the case of human 3D cell cultures, the microscaffold clearly provides better stability and improves the viability of odontoblast-like cells in the core of the organoids, which is in accordance with previous results on cell viability in 3D structures. 39
Discussion
Dental anomalies such as abnormalities in the number of teeth (hypodontia, oligodontia, and anodontia), shape, size, and structure of mineralized tissues (i.e., enamel: AI; dentin: DI and DD; and cementum and alveolar bone), eruption, and resorption may exist as isolated or as a manifestation of syndromic rare disorders.9,40 AI, DI, and DD are rare hereditary anomalies that affect the structure of the tooth hard tissues. All of these conditions result from disruptions in the embryological processes of amelogenesis or dentinogenesis and typically impact both primary and permanent dentition. The phenotypic expression of these genetic diseases, which follow autosomal inheritance patterns, exhibits significant variability. Dentists play a crucial role in the early diagnosis of these disorders and provide guidance to patients regarding measures to preserve affected teeth for an extended period. Affected individuals may experience a diagnostic odyssey averaging 4–5 years or more and encounter difficulties in accessing appropriate care and treatment. Performing molecular and clinical diagnosis of a wide variety of syndromic and isolated AI and DI, the GenoDENT study has revealed that, in 10–20% of the cases, the pathogenicity of the identified genetic variants remains of uncertain significance, and up to 40% of families are in diagnostic wandering. Families with negative results by the targeted NGS approach are then analyzed by WES or even, further, by whole genome sequencing as part of a nationwide plan in France “Plan France Medicine Genomique 2025,” which led to the discovery of new genes involved in odontogenesis. In order to elucidate the pathogenicity of these new genes and identify VUS involved in dental mineralized tissue anomalies, we developed these in vitro 3D organotypic cell cultures using odontoblast and ameloblast-like cells (from mouse and human). This organotypic 3D model is constituted by a coculture of two cell types specific to odontogenesis: odontoblasts (driving dentin morphogenesis)5,41 and ameloblasts (drivers of tooth enamel formation).21,22,42 This study has allowed us to create a low-cost, efficient, and reproducible cell culture system mimicking odontogenesis with the cells responsible for the formation of hard tissues of the tooth, represented by these four characterized cell lines. Compared with traditional 2D cultures, this 3D model offers a more accurate representation of tissue function and enables comprehensive assessment of organoid phenotype, morphology, degradation, mineralization, expression of dentin and/or enamel matrix proteins, and cell differentiation processes.14,17,24
In addition to the coculture of two cell lines of different embryonic origins (epithelial and mesenchymal), this model enabled us to evaluate by confocal and electron microscopy the expression of bilayer proteins during the cellular differentiation of murine and human dental pulp cells and ameloblast-like cells, as well as their secretion of the dentin and enamel matrix in a simplified and reproducible manner. In the case of dentin matrix, we were also able to observe and analyze mineralization of the odontoblastic core of organoids, giving to this model functionality in the study of dental hard tissue abnormalities. In fact, any genotypic change that leads to a relevant change in the phenotype and expression patterns of these cells will be rapidly represented in this coculture system, and the perturbations produced at the protein level will also be easy to observe. The new PLGA microscaffolds were also tested to increase the viability of pulp cells in monocultures recently, 39 and in the search for improved stability and better analysis of human organoids. They were added to the first step of culture in order to form the core of the organoids and then envelop them with ameloblast-like cells. This allowed us not only to test a new biomaterial that can be used for tissue regeneration but also to improve the performance of this model, which, in turn, will allow us to study the genetic perturbations identified in patients by performing functional cellular studies in a 3D in vitro model. Indeed, we are looking forward to interfere these organoids by introducing into each specific cell type (ameloblasts or odontoblasts) a genetic variation of unknown significance (VUS) previously identified patients that have followed a genetic analysis by the GenoDENT NGS panel. In fact, this study conducting translational research on rare diseases through multidisciplinary and transversal approaches has allowed us to develop a new model to perform phenotypic screening using these 3D cellular models. These analyses are an essential support for genetic diagnosis to assess the functional consequences and pathogenicity of these identified genetic variants. The analysis of the effects of VUS on identified molecular markers, specific to amelogenesis and dentinogenesis, will allow us to rectify their functional pathogenicity and their possible involvement in these structural dental anomalies (data to be published). This work is part of the evolution to personalized medicine, as it will allow us to optimize the diagnosis and management of patients referred to our Reference Center for Rare Diseases, CRMR O-Rares of Strasbourg and at the national level to the existing rare diseases networks in France. In order to study the effect of the identified VUS or new genes, ameloblast-like cells or odontoblast-like cells will be mutated by CRISPR/Cas9 for the selected target genes and the identified selected VUS. The mutated cells will then be studied in our coculture system and in a 3D environment, in order to evaluate the phenotype and morphology of the organoids, as well as the expression of dentin and/or enamel matrix proteins, their degradation, and mineralization.
Future experiments are currently being performed on 3D microscaffolds and organoid conception by bioprinting and allowing human odontoblast differentiated cells to maintain their viability over time. 39 Currently, the exploration of bioprinting technologies for the creation of bioprinted and microscaffolded organoids is indeed actively pursued by our group. Bioprinting has significant implications in regenerative medicine, enabling the creation of partial or complete tissue substitutes. Additionally, it offers the opportunity to develop 3D in vitro models that better capture the complexity of diseases, compensating for the limitations of traditional 2D models. This advancement in our research will further enhance the precision and controlled organization of different cell types in a 3D architecture. In fact, the conception of these microscaffolds and their use on the production of new human AM1-hDPSC tooth organoids are parallelly being tested within our team. These results and models may allow us to further functionally study the effect of mutations induced in the human ameloblast-like cells and odontoblast-like cells (data to be published). Through a systematic interpretation of genetic variation data obtained from this concept, we aim to improve diagnostic accuracy and reduce errors. This will lead to the reclassification of variants from class 3 (unknown significance) to class 4 (probably pathogenic) or class 5 (pathogenic), thereby facilitating personalized and individualized diagnoses. This approach is perfectly in line with the construction of a collective vision of biomedical sciences in the field of personalized medicine. Outcomes with this model can contribute to significantly improve the understanding of some rare diseases and their oral manifestations as well as the delivery of a more accurate diagnosis to patients.
Conclusion
We have developed a reproducible in vitro 3D coculture system with ameloblast and odontoblast cell lines where a bilayer structure with cells of distinct embryonic origin, cellular function, and matrix secretion can be observed in a simplified manner. This has facilitated the study of the matrix secretion of mice and human cell populations under differentiation conditions, and new perspectives can be opened on the study of genetic perturbations on each of these two cell types. Moreover, human organoids showed less stability over time than murine organoids, which limits their use in studying the pathogenicity of genetic variants identified in patients with diagnostic impasses. In order to improve the structure and stability of human organoids, PLGA microscaffolds were used to improve the cell viability of odontoblast-like cells in the core of these structures and maintain better in time the cohesion of the two cell populations.
Footnotes
Acknowledgment
The authors specially thank Prof. Thimios Mitsiadis, Foteini Machla, Varvara Platania, Giuseppe Alastra, Laurence Pirenne, and Yves Lutz for their support and technical advice for some of the experiments.
Authors’ Contributions
Review and editing: F.J., V.G., Y.H., Y.A., A.B.-Z., and I.M.B. (equal). Conceptualization: F.J., Y.A., A.B.-Z., and I.M.B. (equal). Writing—original draft: F.J., V.G., T.R., Y.A., A.B.-Z. (equal), and I.M.B. (lead). Formal analysis: F.J., V.G., T.R., S.E., Y.A., A.B.-Z., and I.M.B. (equal). Software, analysis, and results writing: F.J. (lead), T.R., S.E., M.K., N.K., and I.M.B. (equal). Methodology: F.J., V.G., T.R., S.E., M.K., N.K., and I.M.B. (equal).
Disclosure Statement
All authors declare no conflict of interest.
Funding Information
This study was supported by “Impulsion recherche Filière TETE COU” grant 2020 and 2022. Functional validation of VUS in vitro organoids received support from
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.