
Abstract
The current White paper summarizes the discussions and exchange of experiences during the first European Interdisciplinary Summit on Cell-Based ATMPs held in Vienna, Austria, May 02–03, 2013. The meeting was supported by the Research Networking Programme REMEDIC (regenerative medicine) funded by the European Science Foundation and by the British Medical Research Council. To improve the competitiveness of Europe in the field of cell-based Advanced Medicinal Therapy Products (ATMPs), the following key issues were identified during the meeting: removal of national hurdles in the European Union, harmonization of national and subnational differences in Hospital Exemption rules, improved treatment algorithms for reimbursement, better knowledge on the mode of action, predictive preclinical efficacy and safety testing, need for innovative systems for preclinical testing, appropriate product characterization, manufacturing with cost of goods in mind, and appropriate design of clinical trials.
Introduction
T
As defined by the European Commission, Advanced Medicinal Therapy Products (ATMPs) are new medical products based on genes (i.e., recombinant nucleic acids, gene therapy), cells (cell therapy), and tissues (tissue engineering). 1 This white paper focuses on cell-based therapeutics in the form of injectable cells and tissue engineering approaches. The target audience are translational researchers, especially young researchers and those embarking on the drug development process, as well as policy makers.
The biggest challenges and hurdles that need to be overcome to improve the competitiveness of Europe in the field of cell-based ATMPs are
National Hurdles, Harmonization
Both European 2 and US 3 legislative frameworks have laid down rules for complex cell therapies to control the testing, manufacture, marketing, and use of the products. 4 A harmonization of selected rules and requirements across individual European countries is necessary, also between the EMA (European Medicines Agency) and FDA (U.S. Food and Drug Administration), so that the path of product development and associated requirements becomes clear and transparent. Concerted efforts are ongoing in this regard.
A qualified person should be used for all ATMP release, including in the context of clinical trials and for hospital exemptions. More involvement of (hospital) pharmacists is also necessary as it appears that they are not always familiar with this new class of products.
Hospital Exemption, National and Subnational Differences
As the ATMP regulation 5 builds on the Medicine Directive, 6 its scope is limited to products that are intended to be placed on the market in Member States, and which are either prepared industrially or manufactured by a method involving an industrial process. An exemption from the otherwise compulsory centralized licensing procedure is made for those ATMPs that are prepared on a nonroutine basis, according to specific quality standards, and used within the same Member State in a hospital under the exclusive professional responsibility of a medical practitioner (Fig. 1). 5 Relevant community rules related to quality and safety are not to be undermined. This so-called “Hospital Exemption” represents a baseline consensus, from which Member States are called to develop their own regimen.
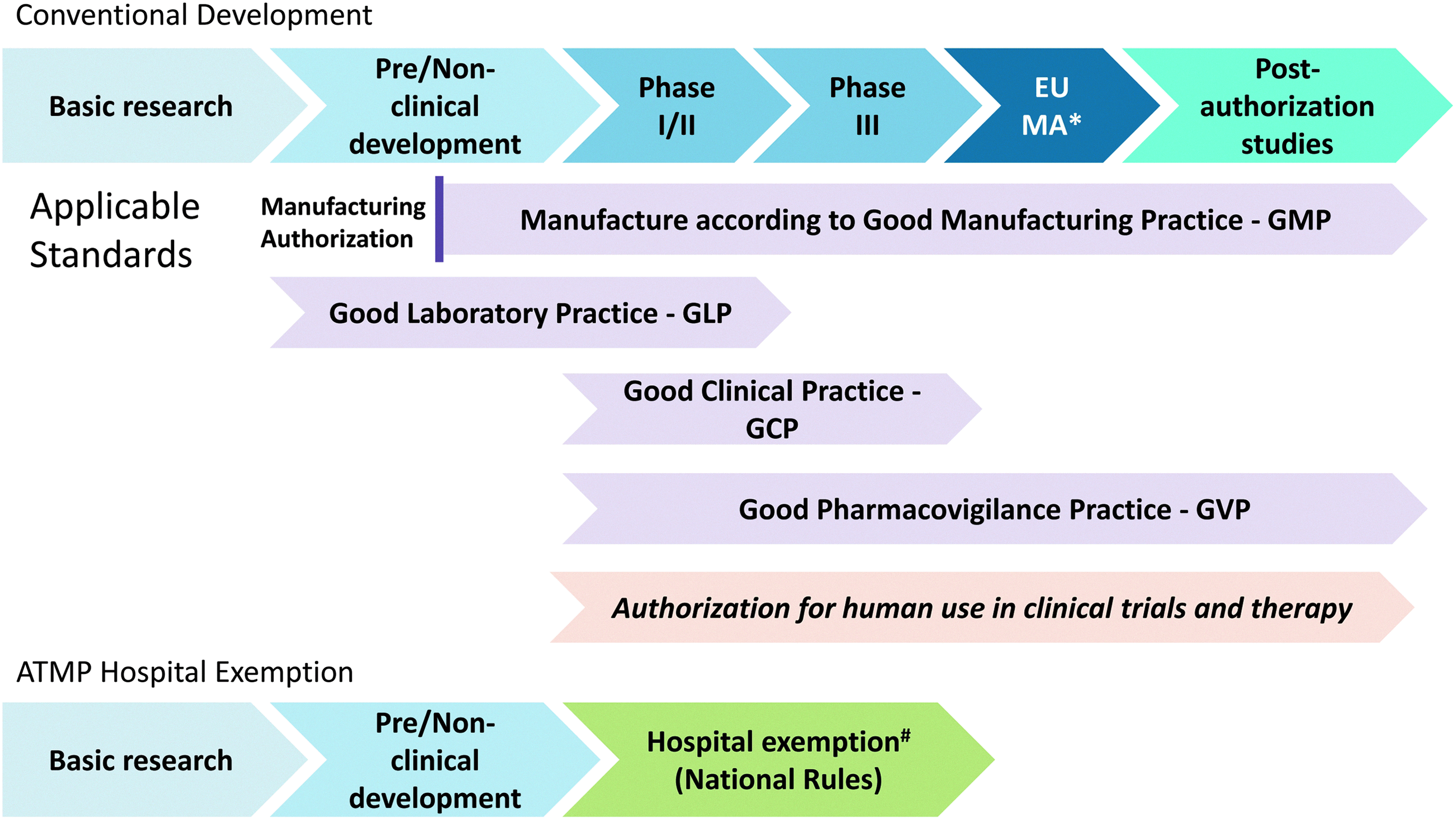
Structured translation of Advanced Medicinal Therapy Products (ATMPs) in Europe *EU Marketing Authorization: Centralized licensure required for ATMPs. #Hospital exemption is limited to Member State. MA, marketing authorization.
The Hospital Exemption is a challenge as many interpret and implement the scope of the Hospital Exemption differently. 7 The field needs clarity about the European rules with regard to hospital exemptions and a uniform implementation of the rules defining when treatments can be done under a Hospital Exemption.
Sufficient room for early exploratory treatments in the hospital setting is required to help the field move forward. However, hospital exemptions should not be used for the sake of local hospitals to treat patients routinely with cell-based products or to accumulate clinical data for a certain product in countries that do not implement strict Hospital Exemption rules. Several countries are already quite clear and transparent in this regard, others seem to remain undecided. In the long run, efficacious and safety data need to be generated and that is only possible through rigorous clinical trials.
Reimbursement
Positioning cell-based treatments in daily clinical practice should be addressed early in development. Early-on incorporation of approaches allowing to define appropriate patient target groups (patient stratification) and responders to treatment (e.g., using biomarkers) is critical toward a personalized medicine approach, in particular, for this new class of treatments.
Collection of patient data in an attempt to properly position the treatments in daily clinical practice is an important approach for successful reimbursement discussions in the future. This can lead to new or updated treatment algorithms, and will thus be of help for the identification of well-targeted patient groups in the context of reimbursement.
Knowledge on the Mode of Action
Although it is clear that living cells are much more complex systems than chemical drugs, it is required to better understand their main actions and attributes responsible for the intended therapeutic effect. For example, it is often not clear whether the therapeutic efficacy of a cell-based ATMP is based on a progenitor or a nonprogenitor function, or when multiple components are being used, which of those appear fundamental for the intended activity. 8 This understanding is expected to allow for a more targeted optimization of the cell-based product. 9 To address this goal, novel and multidisciplinary approaches may be needed.
Part of the mode of action is also the characterization of the functionality of the cells/tissues derived from the cell-based product, when applicable. For instance, functional cardiomyocytes are expected for regeneration of infarcted myocardium or functional neurons expected for regenerated spinal cord.
When using human cells in animal models, immunological reactions (rejection) of the host might impair the behavior of the donor cells (e.g., engraftment, growth, and proliferation).10–12 Therefore, the use of homologous products may be advisable if practically feasible, showing the concept to work in the species tested, and providing clarification of the mechanism. One has to take into consideration particular aspects of the model that might/might not differ from those in humans. The models need to be justified, their relevance to mimic humans needs to be shown, and the aspects relevant for translation into humans also need to be defined.
Predictive Preclinical Efficacy and Safety Testing
As for any new medicinal product, the development and approval of cell-based medicinal products need to be based on nonclinical data supportive of the mode of action and potential therapeutic action, and on appropriate safety attributes. 9 Due to their complexity and the human origin of most cell-based ATMPs, the existing preclinical models are sometimes irrelevant for prediction of effects in human patients, and need to be adapted on a case-based manner. While the safe use of any new cell-based product needs to be predicted before entrance into human experimental or therapeutic use, the strategies for data generation and the decision on which data are needed should be planned well in advance, taking into consideration the particular aspects of each product. The use of relevant experimental models is of outstanding importance, as only those will be able to appropriately support the efficacy and safety of the product. Data generated in irrelevant models are a concern, as they could give a false sense of risk or safety that may heavily impact on the appropriate use of the product.
With regard to the safety assessment of a cell-based product, general studies on safety pharmacology, repeated dose toxicity (addressing general toxicity), reproductive toxicity, and carcinogenicity are in principle needed, similar to conventional drugs. However, the experimental strategies and study design to generate such data may need adaptation to the specific nature and potential safety concerns of such living, biological products. Data and information collected from mode of action studies may also be helpful to establish safety attributes and to design the safety studies. Since a single dose of a cell-based product may lead to a persistent or long-lasting exposure, its biodistribution, persistence, and potential for tumor formation need to be addressed and anticipated. The extent of biodistribution, potential for ectopic engraftment, and the persistence at intended and/or unintended sites of action will dictate the extent and the type of safety studies further needed (Fig. 2).

Relevant aspects for risk prediction of cell-based medicinal products (CBMP), to be considered when planning the preclinical studies, as outlined by the Guideline on Human Cell-Based Medicinal Products. 9 SOA, site of administration.
It is important to keep in mind that for the development of any cell-based product, the characteristics of the cell product itself will dictate the type of safety concerns associated, and those that are expected to be anticipated and tested, based on the existing knowledge about the product (cell) type. This has formed the basis for the so-called “risk-based approach” called for by the Committee of Advanced Therapies of the EMA (the CAT), which has issued a guideline 13 proposing that, for any new ATMP, risks are anticipated based on the knowledge of the product (meaning that it will be science driven and time evolving). The design for safety and efficacy studies, nonclinical or clinical, should take these risks into consideration, in addition to the quality aspects.
An example for a risk-based approach is the preclinical development of ChondroCelect™, a tissue-engineered product for repair of damage to the articular cartilage in the knee. The classical development plan was not entirely followed, and the options to omit some studies (safety pharmacology, genotoxicity, in vivo carcinogenicity) and to include some cell therapy relevant studies (like cell senescence) were taken. A product-based approach was thus adopted, with appropriate justifications. 14
As for proof-of-concept studies, the animal model used for in vivo studies needs to show relevance with regard to the human situation. While rodents are easy to use, large animals may also be needed, depending on the product type and indication (e.g., horses or goats for cartilage repair or pigs for spinal cord injury). However, large animals are difficult to use for safety purposes, and their use is normally more common for proof-of-concept studies. Nevertheless, inclusion of safety endpoints in proof-of-concept studies, whenever possible, will make those more informative and will contribute to the reduction of animal use according to the 3R policy.
While the use of homologous products might be the most helpful and meaningful for the prediction of any efficacy and/or safety concern associated with the cell-based product concept, it is usually not accepted by regulatory authorities that a human cell-based product is developed based on conceptual data only, without actual testing of the clinical candidate. This means that the genuine cell-based medicinal product needs also be tested, which, being of human origin, will lead to immunological reactions in animal species. Immunosuppressed animals (rodents) are mostly used to overcome this problem in safety testing. To allow for a good understanding of the animal-specific features, and in the ideal situation, it is advisable that the clinical candidate is tested in immunosuppressed animals in conjunction to the homologous product in immunocompetent animals. The inclusion of safety endpoints in the proof-of-concept study may overcome the need for separate safety studies with the homologous product. Taking these aspects into consideration is obviously a matter of careful early planning, starting with the proof-of-concept.
It is expected that a science-driven, case- and risk-based approach for development of cell-based products will improve characterization and facilitate early planning of necessary study packages for successful marketing (Fig. 2).
Need for Innovative Systems for Preclinical Testing
For many cell-based products, it is possible to anticipate their human efficacy and safety based on the knowledge associated with product characteristics, and the outcome of their testing (or of testing of homologous products) in animal species. However, there are cases in which animal efficacy and safety data are difficult to interpret and extrapolate to humans, for example, for cancer immunotherapy, where efficacious products in rodents have shown highly disappointing results when entering clinical trials.
When animal models are judged as not being useful or misleading, alternative approaches are commonly considered as rescue options, using in vitro systems that desirably are based on human systems (cells/tissues in culture). When the preclinical development of a product is based exclusively on in vitro data, the transfer into humans for the first clinical trials is made with an extra-cautious approach, but still cases do exist, and the unavailability of appropriate animal models should not be a reason for blocking a development plan, particularly in therapeutic areas of high need.
The advances in the field of cell-based therapies led to increased knowledge about different cell-type attributes, identification markers, methodologies for (sub)cellular access, identification, characterization, growth, isolation, and culturing. Efforts have also been made toward achieving coculturing cell systems, three-dimensional (3D) cultures, and organotypic cultures. While the field is still far from being ready for full use, an enormous research investment is being put in the creation of humanized tissue/organ-like systems, which could be used for the testing of compounds directly in human-derived material. In case of success, those strategies, once integrated, may allow translational research concepts to move from the traditional animal into human path toward an in vitro into in vivo human path.
The need for such alternative in vitro systems, based on human cells, allowing appropriate and reliable testing of the main safety aspects associated with new pharmaceuticals is paramount. For cell-based products, this is even more relevant given the specific nature and sometimes questionable relevance of the animal models being used. In an ideal situation, it is expected that systems like the human on a chip, where multiple organotypic constructions are kept connected by microfluidic circulation, would allow the understanding of the potential cell products to express their intended and unintended properties, covering tissue access, tissue engraftment, ectopic engraftment, cell persistence, senescence, degeneration, cell-derived product identification, etc. If appropriately collected and interpreted, data generated in these systems, based on well-formulated questions (e.g., the risk-based approach), might allow a careful introduction of such products in humans, with appropriate tools for cell tracking (e.g., imaging) and even cell suicidality in case of need.
Multiple initiatives in the European arena and worldwide (see, e.g., http://wyss.harvard.edu/viewpage/461/) are currently being carried out to put these possibilities into reality for the sake of improved success in drug development and human health.
Product Characterization and Product Potency
Product characterization and analysis of product potency for ATMPs may be of higher complexity than for other biochemically derived biologicals, 15 but nevertheless are of high importance due to potential manufacturing process variability and the living nature of the product. 9 Current approaches used in cell therapy product manufacturing processes often include biologically variable starting materials, and the criticality of a wide range of process steps for the quality of the final product in terms of impact on the living cells and their behavior, and ultimately, product potency is often unclear. Early efforts in product characterization and development of potency assays (combined with a structured process development approach) allow fact-based evaluation on product consistency when changes and improvements are made to the manufacturing process. Conceptually, this provides a type of insurance premium to enable effective continuation of the project, even at times when important unexpected process outcomes are experienced, or changes in reagents or to the manufacturing process become necessary: the fact base allows charting a path forward. A further important aspect of product characterization is the development of a scientific rationale for the dose to be applied to the patient. 16
Manufacturing with Cost of Goods in Mind
Whereas in early-stage clinical trials, the emphasis of many companies is exclusively on manufacturing products according to specifications and under Good Manufacturing Practices conditions; later the focus will promptly shift to scale-up and achieving acceptable cost of goods.17,18 A clear understanding of the target indication, its value range, and estimation of patient cell dosages will be required to come to an evaluation of whether the initial product platform will be able to deliver commercially acceptable costs. In most cases, early attention is recommended to implement scale-up activities. From such an integrative perspective, one can carefully select steps to either achieve scale-up immediately or to allow easier scale-up at a later phase. A clear path needs to be charted on how to deal with the scale-up question early on, even though a delay may be required due to lack of funds in earlier clinical phases. Investors will be keen to have an in-depth understanding on how scale-up can be achieved as this is currently seen as one of the most important hurdles of commercial success for cell-based ATMPs.
Clinical Trials: Design and Outcomes
Clinical trials are necessary to allow the assessment of the safety and efficacy of new cell-based ATMPs. 19 It involves exploratory trials and confirmatory trials. Typically, a phase I/IIa study looking at the safety and feasibility of the treatment, with some indications of efficacy. Already in this phase, an effort should be made to identify the type of patients who are responsive to these treatments, allowing for potential patient stratification in subsequent trials.
Randomized controlled phase II studies are mandatory to assess dose finding, treatment efficacy, and safety. (Multicenter) phase III studies are needed for definitive testing before market authorization or employment as standard treatment (Fig. 1).
After market authorization, it is suggested that the collection of postmarketing real-life patient data in daily clinical practice is more useful and relevant over running many smaller, confirmatory trials in different patient subpopulations. The rationale behind this approach is to better define the right product for the right patient at the right time, information that may also be used for adaptation and fine-tuning of the reimbursement criteria. All patients treated in clinical trials and in later standard treatment should be monitored for long-term safety and efficacy (Fig. 1). In the ideal situation, this long-term vigilance monitoring should include data on quality of life and treatment costs.
More adapted trial designs may be appropriate as many indications for the cell-based ATMPs are for niche patients, potentially orphan drug indications.
Long-term follow-up is of importance, in particular, for allogeneic cell-based treatments. Development of methodologies to trace cells over time appears important, also in view of long-term safety.
Conclusions
The clarification of the mode of action of any cell-based ATMP, as for any other medicinal product, should ideally be undertaken early in the discovery phase, where basic scientists in academia and small enterprises are mostly the driving forces. To facilitate the progress of their concept through development as a candidate ATMP, researchers need to be made aware of how relevant it is that the mode of action is defined, and that the knowledge generated on the mode of action will highly impact on the success of the product development. Because the majority of stakeholders developing ATMPs are academic groups and small companies, the capacity to navigate the regulatory process is limited, and therefore, direct interaction with regulators is recommended to become more familiar with the regulation. 19
ATMPs are a new class of products, and their development has been a challenging process. There are numerous potential applications, in particular, in the field of regenerative medicine. It brings with it the development of new enabling technologies for manufacturing approaches, including 3D bioprinting, new bioreactors and biosensors, microfluidics, and noninvasive imaging to name a few. This implies a high level of interdisciplinarity and a critical mass of frontline researchers concentrated in clusters. Europe, with an accessible high-quality medical care system and a booming biotech industry, is well placed to become a leader in this field. Aside from the political need for removal of national hurdles and for harmonization of regulatory differences in the European Union, more close collaboration between academia, industry, and regulators will be essential to move the field forward and to close the translational gap in Europe.
Footnotes
Disclosure Statement
W. Dalemans, A. Vos, and R.T.A. Janssen are employees of TiGenix, Pharmacell, and Scalable Life Sciences, respectively.