
Abstract
Surgeons have used cryopreserved vascular allografts successfully for many years to treat arterial occlusive disease and to repair arterial aneurysms. Vascular allografts demonstrate high patency rates but contain viable cells, which may evoke a rejection response following implantation. Removing the cells could prevent such a response and negate the need for cryopreservation and ultra-low temperature storage.
The objectives of the study were to characterize human common femoral arteries and develop a decellularization protocol with a view to the generation of biocompatible and biomechanically functional vascular grafts for use in vascular bypass and arteriovenous access. The arteries were decellularized by subjecting the tissue to a single freeze-thaw cycle followed by sequential incubation in hypotonic tris buffer and low concentration sodium dodecyl sulphate. Each artery was disinfected using 0.1% (v/v) peracetic acid.
Histological analysis demonstrated a lack of cells following decellularization and confirmed the integrity of the tissue histioarchitecture and retention of major structural proteins. There was a >95% reduction in DNA levels. The acellular tissues and extracts were not cytotoxic to either mouse 3T3 or baby hamster kidney cells. Biomechanical properties were determined by burst pressure, compliance, and tensile tests, which confirmed the retention of biomechanical properties following decellularization.
In conclusion the study has developed a suitable protocol for the removal of cells from human common femoral arteries without adversely affecting the biochemical or biomechanical properties. These properties indicate the potential use for acellular human common femoral arteries for vascular bypass or arteriovenous access.
Introduction
Vascular allografts, mainly veins, have been used as an alternative to synthetic grafts for many years to treat arterial occlusive disease and to repair arterial defects.4,5 The popularity of allografts has risen and fallen many times over the years. 5 Recently, improvements in cryopreservation techniques have renewed interest in vascular allografts. Infection of a synthetic graft is the primary indication for the use of an allograft. 5 Allografts have potential advantages over synthetic grafts that include reduced incidence of infection and increased patency rates since up to 80% of the endothelial cells lining allografts have been shown to be viable at the time of implantation.6,7 However, the clinical use of allografts may lead to allosensitization. 7 The expression of class I and II major histocompatability complex antigens by viable endothelial cells within vein allografts has been reported to initiate an immune response when they were used for hemodialysis access. 7 Studies have suggested that cold-stored grafts with no viable cells may not be immunogenic and that cold storage for 4 weeks in physiological buffer does not adversely affect the extracellular matrix or biomechanical properties. 8 Early strategies to produce alternatives to allogeneic vessels for bypass and arteriovenous access have included seeding autologous or allogeneic cells onto synthetic nondegradable scaffolds. 9 These grafts were shown to have sufficient mechanical strength but failed to achieve clinical use. More recent strategies involve the use of natural extracellular components such as collagen or biodegradable polymers to fabricate scaffolds, which are subsequently seeded with autologous cells; the use of mature cell sheets has also been pioneered as a potential vascular graft.10–13 The major limitations of these approaches are a lack of mechanical strength and lengthy production time. 10
The use of decellularized tissues has the potential to overcome limitations of conventional tissue engineering approaches and is an attractive technique for scaffold preparation, as the resulting material can potentially retain the architecture of the original tissue. Decellularized tissues are composed of natural extracellular matrix. The goal of any decellularization process is to remove all cellular components thereby reducing the immunogenicity of the tissue while preserving the original biological and mechanical properties.14,15 Decellularized tissues may subsequently be seeded with cells or implanted as an acellular scaffold.11,16 An acellular allogeneic graft would provide many benefits over a cryopreserved graft including reduced immunogenicity and off-the-shelf availability. Umbilical and saphenous veins have most widely been decellularized but recently the interest has been directed toward umbilical arteries. 17 The protocols used to decellularize allogeneic vessels commonly involve the use of sodium dodecyl sulfate (SDS),18,19 a combination of (3-[(3-cholamidopropyl) dimethylammonio]-1-propane-sulfonate) (CHAPS) and SDS, 17 osmotic stress, 20 detergents and endothelial growth medium, 17 and the use of ethanol to aid lipid extraction. 19 SDS (1.8 mM)/CHAPS (8 mM) treated arteries resulted in vessels with reduced elastin content and a diminished compliance at low pressures. 17 The use of arteries is a logical choice for vascular access and bypass applications due to their superior biomechanical and handling properties. The most important characteristic of any vascular graft is adequate initial strength; this requirement makes decellularization approaches attractive. However, there is currently no decellularized human vascular graft in clinical use. The ideal acellular allogeneic vascular graft would be devoid of cells and genetic information while retaining the extracellular matrix components and histioarchitecture, and it is nonthrombogenic, and most importantly, it has biomechanical properties similar to the artery being replaced or repaired. The aim of this study was to develop a decellularization process for human common femoral arteries, which retained the structure and biomechanical properties of native arteries while being free from cells and DNA.
Materials and Methods
Tissue procurement
Allogeneic common femoral arteries were obtained through Tissue Services (NHS Blood and Transplant) part of the NHS, United Kingdom. The study was carried out under ethical committee approval and all the tissues were obtained following informed consent. Arteries were obtained from cadavers within 48 h of death. Donors were serologically negative for human immunodeficiency virus, hepatitis virus type B, hepatitis virus type C, and syphilis. In the laboratory the adventitia was removed by blunt dissection to aid the decellularization process and each artery was washed three times in Dulbecco's phosphate-buffered saline (DPBS; Oxoid) containing 0.1% (w/v) ethylene diamine tetraacetic acid (EDTA; VWR) to remove excess blood. Tissues were then stored at −80°C on DPBS-moistened filter paper for future use. All vessels were 15–26 cm in length and varied in internal diameter from 3 to 7 mm. The arteries used throughout the study were of continuous length, beginning with the external iliac artery and continuing distally to the superficial femoral and popliteal artery; throughout the manuscript they will be referred to as common femoral arteries.
Decellularization
Decellularization was performed based on the procedure described by Booth et al. 21 Arteries were processed individually in 200 mL of solution. Arteries were subjected to two cycles of hypotonic buffer (10 mM tris; pH 8.0) at 4°C for 24 h and hypotonic buffer containing 0.1% (w/v) SDS (Sigma) at 37°C for 24 h (pH 7.4) with agitation in the presence of protease inhibitors (Aprotinin, 10 KIU·mL−1; Trasylol and 0.1% [w/v] EDTA; VWR). Arteries were washed in DPBS three times for 30 min with agitation. Following washing, arteries were incubated in DNAase (50 U·mL−1; Sigma) and RNAase (1 U·mL−1; Sigma) in buffer (50 mM tris-HCl, 10 mM MgCl2, 50 μg·mL−1 bovine serum albumin [Sigma] at pH 7.5) for 3 h at 37°C with gentle agitation. Each artery was then washed in hypertonic buffer (1.5 M NaCl in 0.05 M tris-HCl, pH 7.6) for 24 h at 37°C with agitation. A decontamination step, consisting of incubating the arteries in 0.1% (v/v) peracetic acid (Sigma) in DPBS for 4 h, was then incorporated to achieve surface disinfection of the tissue.22,23 Finally, the arteries were washed three times in DPBS at 4°C for 30 min each with agitation followed by two 24-h washes. Acellular arteries were compared with native arteries to determine the effects of the decellularization process on the biological and biomechanical properties of the tissue.
Tissue sampling for biological evaluation
A total of six acellular arteries and six native allogeneic common femoral arteries [15–26 cm in length] were compared by histology, immunohistochemistry biochemical assays, scanning electron microscopy (SEM), and biocompatibility. Segments were taken from either the end (n=3) or the central regions (n=3) of each artery for each of the analyses. Histological analyses were carried out on a minimum of 10 sections per segment.
Histology
Native and acellular arterial segments were fixed using 10% (v/v) neutral-buffered formalin for 1 h and then dehydrated using an automated process before being embedding into paraffin wax. Hematoxylin and eosin (H&E; Raymond A Lamb Ltd) staining was used to evaluate tissue histioarchitecture. Alcian blue (Raymond A Lamb Ltd) staining (1%; w/v pH 2.5) was used to localize glycosaminoglycan (GAG) content. Sirius red Miller's stain (Raymond A Lamb) was used to visualize collagen and elastin distribution and orientation. Cell nuclei were visualized using DAPI dye (Invitrogen). Sections were viewed using an Olympus BX 51 microscope. Images were captured digitally using an Olympus XC50 camera and the software used was Olympus soft imaging solutions.
Immunohistochemistry
Segments of native and acellular arteries were fixed using a zinc fixative for 24 h at room temperature (BD). Monoclonal antibodies against collagen type I (1:50), collagen type IV (1:50), fibronectin (1:50), laminin (1:20), and Von Willebrand's factor (1:20; Dako) were used to establish collagen distribution and the presence of basement membrane proteins. Immunolabeling was carried out using the Envision kit (Dako), an indirect streptavidin-horseradish peroxidase immunoperoxidase method. Tris-buffered saline (pH 7.4) was used throughout as the diluent and wash buffer. Nonspecific background staining was prevented by blocking with 10% (v/v) rabbit serum (Dako) and 3% (v/v) hydrogen peroxide (Sigma). Omission of the primary antibody served as the negative control. Fresh human skin was used as a positive control tissue and isotype control antibodies (IgG1 and IgG2; Dako) were used to verify antibody specificity. Following staining, all images were captured digitally and qualitatively assessed.
Scanning electron microscopy
Segments of native and acellular arteries were fixed with 2.5% (w/v) glutaraldehyde/DPBS, postfixed and stained with 1% (w/v) osmium tetroxide, and subsequently dehydrated using an ascending alcohol series. The samples were critical point dried using a Polaron E3000 critical point drying apparatus (Aztech trading) using liquid carbon dioxide (CO2) as the transition fluid. Specimens were mounted using carbon cement and coated with platinum using a Polaron E5300 freeze dryer, sputter coating unit. Specimens were then observed and imaged using a QUANTA 200 field emission gun scanning electron microscope (FEI) to evaluate the structure of native and acellular arteries. All images were captured digitally.
Biochemical assays
Hydroxyproline assay
Prior to performing the hydroxyproline assay, segments of native and acellular arteries were lyophilized to a constant weight before being hydrolyzed by incubation with 6 M hydrochloric acid for 4 h at 120°C and neutralized using sodium hydroxide. The procedure adopted was based on the method described by Edwards and O'Brien. 24 The concentration of hydroxyproline was determined by interpolation from a trans-4-hydroxy-L-proline standard curve. The total collagen content was calculated by using a hydroxyproline to collagen ratio of 1:7.14. 25
Denatured collagen assay
Prior to performing the hydroxyproline assay, segments of native and acellular arteries were lyophilized to a constant weight before being digested using α-chymotrypsin (5 mg·mL−1; Sigma) for 24 h at 30°C in a 0.1 M Tris buffer containing 2.5 mM calcium chloride. This method was based on the procedure described by Bank et al. 26 The concentration of hydroxyproline was determined as described previously.
Sulfated sugar assay
The sulfated sugar content of acid-hydrolyzed segments (6 M HCl at 120°C for 4 h) was determined using dimethylene blue (Sigma) according to Farndale et al. 27 The GAG content was determined by extrapolating values from a standard curve of chondroitin sulfate B (Sigma).
DNA extraction
Total DNA was extracted using a DNA isolation kit for tissues (Qiagen). Briefly, 100 mg (wet weight) of native and acellular arteries was digested using a Proteinase K solution. Following this, an RNAase solution was applied to digest any RNA present within the samples. Samples were loaded onto spin columns and washed to remove protein and other potential contaminating matter. The DNA was eluted into a sterile Eppendorf and quantified using a NanoDrop spectrophotometer at 260 nm (Thermo Fisher Scientific).
Cell culture
3T3 murine fibroblasts (Health Protection Agency) were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% (v/v) fetal calf serum, 100 U·mL−1 penicillin, 100 μg·mL−1 streptomycin, and 2 mM L-glutamine (Lonza) at 37°C in 5% (v/v) CO2 in air; the medium was exchanged every 2 days.
BHK baby hamster kidney cells (Health Protection Agency) were cultured in Glasgow's modified Eagle's medium (GMEM) containing 5% (v/v) fetal calf serum, 100 U·mL−1 penicillin, 100 μg·mL−1 streptomycin, 2 mM L-glutamine (Lonza), and 10% (w/v) tryptose phosphate broth (Oxoid) at 37°C in 5% (v/v) CO2 in air; the medium was exchanged every 2 days.
Contact cytotoxicity assay
Samples of acellular arteries measuring 5×5 mm were attached to the center of six-well tissue culture plates using type I collagen extracted from rat tail tendons. Cyanoacrylate glue and collagen alone in duplicate wells were used as positive and negative controls, respectively. Mouse fibroblasts and baby hamster kidney cells were seeded into each well at a density that would allow confluency after 48 h in their standard culture medium. Plates were incubated at 37°C in 5% (v/v) CO2 in air for 48 h. Following incubation, the culture medium was carefully aspirated before the wells were washed with DPBS containing calcium and magnesium and fixed with 10% (v/v) neutral-buffered formalin (Raymond A Lamb) for 10 min. The wells were then stained with Giemsa solution (Merck R66 formulation; VWR) for 5 min. The wells were then repeatedly rinsed with distilled water until clear and they were air dried. The dry plates were then examined by phase-contrast microscopy using an inverted Olympus IX 71 microscope to view any changes in cell morphology and confluency. All images were captured digitally.
Extract cytotoxicity assay
Segments of acellular arteries were finely minced and left to incubate in DMEM or GMEM (3 mL·mg−1) for 72 h at 37°C with agitation. Following incubation, the mixture was centrifuged (500 g, 15 min) and the supernatant was collected. Mouse fibroblasts and baby hamster kidney cells were seeded onto 96-well plates at a density that would allow confluency after 24 h in standard culture medium. Plates were incubated at 37°C in 5% (v/v) CO2 in air. Following incubation, the culture medium was removed and 100 μL of fresh culture medium was added. To this, 100 μL of test extracts was added to the seeded wells and further incubated for 24 h. Positive controls (40%; v/v) of dimethyl sulfoxide (Sigma) in DMEM and a negative control (DMEM) were included. The cell viability (relative cellular adenosine-5-triphosphate [ATP] content) was then determined using the ATPLite-M assay (Packard).
Tissue sampling for biomechanical evaluation
A total of six acellular arteries and six native allogeneic common femoral arteries (15–26 cm in length) were compared by burst pressure, suture retention, low strain rate failure, and compliance testing. Samples were taken from either the end (n=3) or the central regions (n=3) of each artery for each of the tests. Suture retention and low strain rate failure testing were carried out on samples of arteries measuring 3×20 mm dissected parallel to both the circumferential and axial directions.
Burst pressure testing
Samples of native and acellular arteries measuring 5 cm in length were subjected to increasing internal pressure. Each vessel was in turn mounted into a custom-built burst pressure rig and the open end was ligated. Each vessel was filled with DPBS and checked for leaks. The internal fluid pressure was rapidly increased to 3750 in 50 mmHg increments. The maximum pressure that was able to withstood by each artery before bursting was recorded.
Low strain rate failure testing
Native and acellular arteries were subjected to low strain rate uniaxial loading to failure. Prior to testing, the thickness of the samples was measured at three points along their long axis using a gauge with a resolution of 0.01 mm (Mitutoyo), and their average thickness was recorded. Subsequently, the samples were mounted onto a purpose-built holder. The holder was supported by a removable bracket that allowed alignment of the two holder grips, defined the gauge length of the specimen (3 mm), and ensured that no load was imposed onto the specimen until the start of the test. Once the test sample was clamped onto the holder, the holder with supporting bracket was secured to an Instron tensile testing machine and the bracket was removed. The samples were then stretched to failure at a rate of 10 mm·min−1.
To obtain an accurate measure of the tissue gauge length, a specimen preloading of 0.02 N was prescribed. Therefore, zero extension was taken at the point where a load of 0.02 N was detected. The final gauge length of the specimen was calculated as the initial gauge length plus the extension that was needed to produce the prescribed preloading. Failure was taken to occur when the first decrease in load was detected during extension. Failures occurring away from the center of the sample were excluded. The recorded load and extension data were converted to engineering stress and engineering strain. The calculated stress–strain responses of the test samples were averaged over the number of samples in each group (n=6) using a mathematical analysis software package (Origin v6.0; Microcal Software Inc). The stress–strain behavior of each sample was analyzed by means of six parameters. These have been described elsewhere and included the elastin (EL-E) and collagen (Col-E) phase slope, transition stress (σtrans) and strain (ɛtrans), and ultimate tensile strength (σUTS) and failure strain (ɛUTS).
Suture retention testing
Samples of native (n=6) and acellular (n=6) arteries measuring 1.5×20 mm were dissected parallel to the circumferential and axial directions and subjected to suture retention testing. Prior to testing, the thickness of the samples was measured at three points along their long axis using a gauge with a resolution of 0.01 mm (Mitutoyo), and their average thickness was recorded. Subsequently, the samples were mounted onto the bottom half of a purpose-built holder. A 4–0 Prolene suture (Ethicon) was placed into the free end of the tissue; this was attached to the testing rig. The test parameters from the tensile testing were used and the maximum force withstood before the suture tore out was recorded.
Compliance testing
Samples of native and acellular arteries measuring 5 cm in length were subjected to increasing internal pressure. Each vessel was in turn mounted into a custom-built burst pressure rig and the open end was ligated. Each vessel was filled with fluid and checked for leaks. The internal fluid pressure was increased in increments of 100 mmHg. The horizontal and vertical displacement of the artery was determined by taking a picture of points drawn on the artery prior to inflation. The distance between each point was determined using Image Pro Plus version 4.5.1 (Media Cybernetics) and a graticule.
Disinfection validation
The validity of 0.1% (v/v) peracetic acid as a chemical sterilant was determined using Bacillus atrophaeus spores. A known number of spores were added to the lumen of acellular arteries; each end was ligated and immersed in 200 mL of 0.1% (v/v) peracetic acid at 27°C. Following incubation, each vessel was macerated and the suspension was diluted using DPBS and the number of colony forming units was determined by plating out onto dextrose tryptone agar. This was carried out at 30 min intervals over a 4 h period for each of six acellular arteries.
Data analysis
All numerical data were analyzed using Microsoft Excel 2002. All numerical values are shown as mean values ±95% confidence limits. Statistical significance was assessed by one-way analysis of variance (ANOVA), using the mean value of each sample group, and the minimum significant difference between individual group means was calculated by the T-method, and p-values of <0.05 were considered significant.
Results
Histological analysis
Native arteries
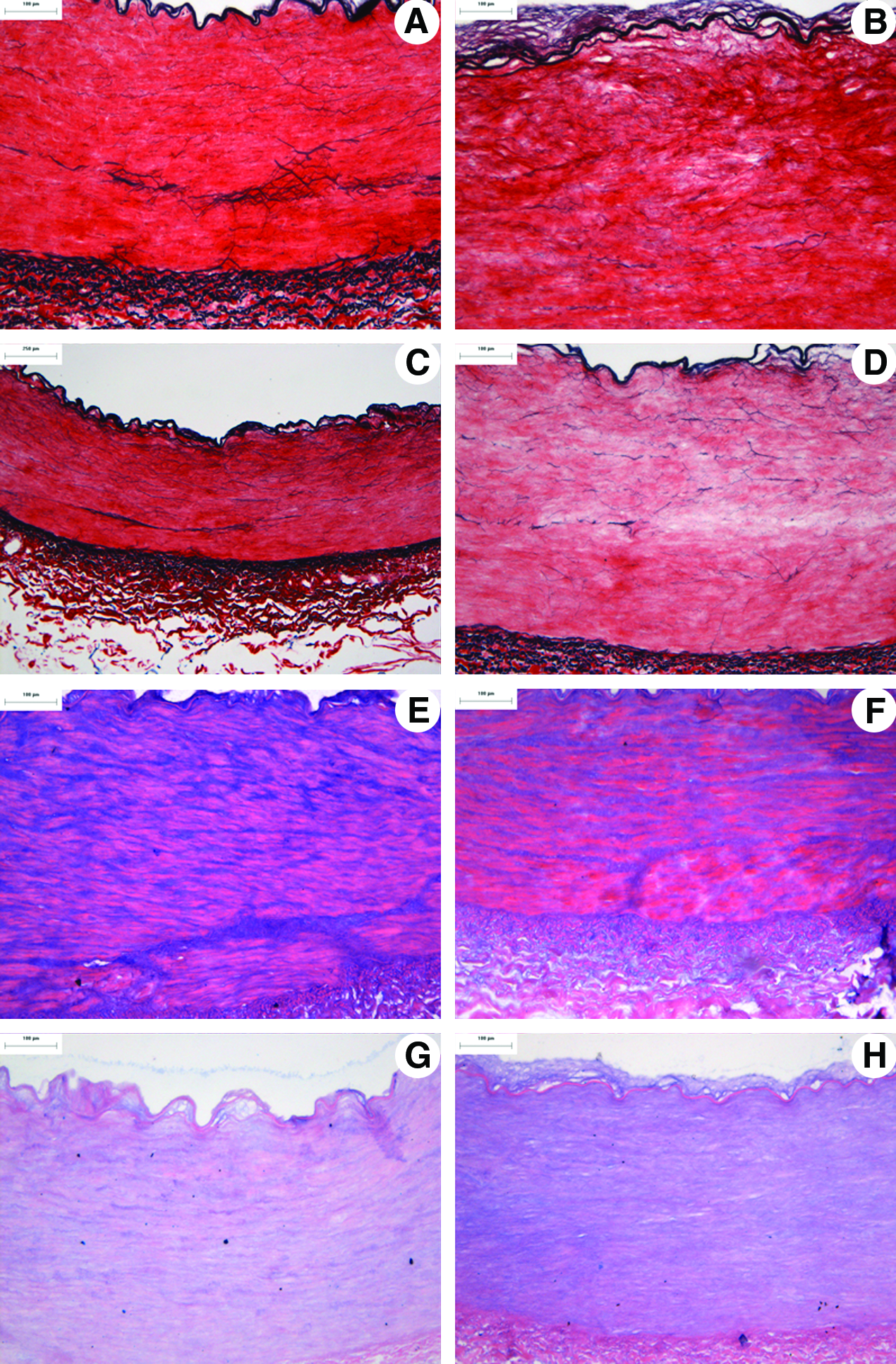
H&E and 4′,6-diamidino-2-phenylindole (DAPI) staining demonstrated the presence of cells throughout the common femoral arteries (Fig. 1). The vessel demonstrated a typical trilaminar arterial structure (Fig. 1). The intima is the inner lining of the artery, consisting of endothelium and a relatively thin layer of supporting connective tissue. The tunica media is the middle muscular and elastic layer, containing of smooth muscle and elastic fibers. The tunica adventitia is the most superficial layer comprised of dense irregular connective tissue with collagen fibers running in all directions. Miller's Sirius red elastin staining revealed collagen and elastin fibrils that are present within the arteries; these were circumferentially orientated (Fig. 2). Longitudinal sections demonstrated collagen and elastin fibers running axially, perpendicular to those observed when transverse sections were stained (data not shown). Alcian blue staining revealed only a small amount of GAGs to be present within the arteries (Fig. 2). GAG concentration was also found to be highest around endothelial cells and the tunica intima (Fig. 2).
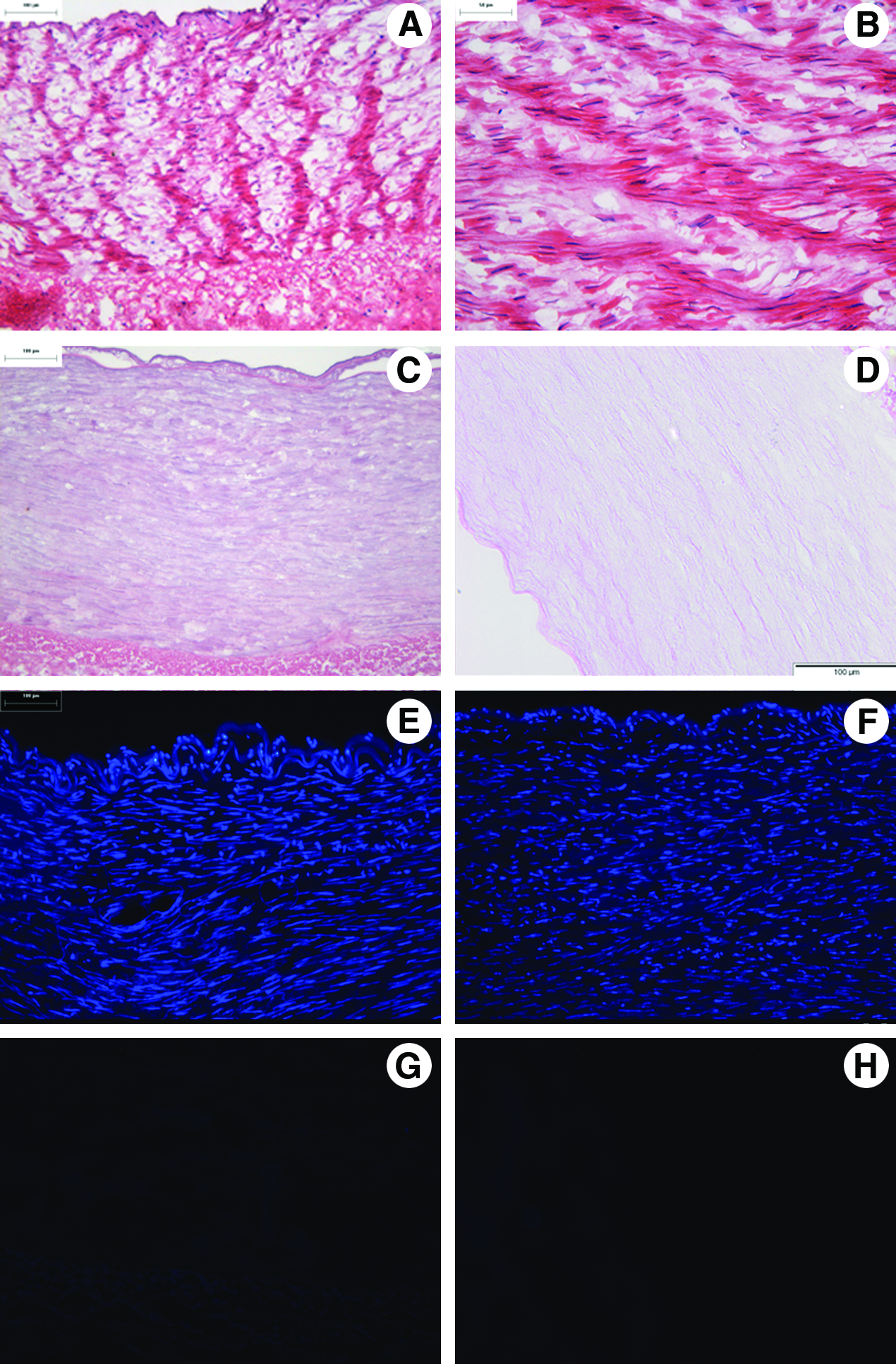
Representative histological images from the central region of fresh
Representative histological images from the central region of fresh
Acellular arteries
H&E and DAPI staining of the acellular arteries showed an absence of visible whole cells and cell nuclei (Fig. 1). Collagen stains (Sirius red and H&E) gave evidence of the retention of the major histioarchitecture following decellularization (Fig. 2). Miller's Sirius red staining demonstrated the retention of elastin fibers following treatment (Fig. 2). Most importantly the images demonstrated the retention of the internal elastic lamella following decellularization (Fig. 2). The histology indicated that there was no change in the distribution of GAGs within acellular arteries compared with native samples (Fig. 2).
Immunohistochemistry
Positive control tissues and isotype control antibodies allowed verification of the antibody panel used: collagen type I, IV, fibronectin, laminin, and Von Willebrand's factor (data not shown).
Native arteries
The native arteries stained positively for collagen I, IV, fibronectin, laminin, and Von Willebrand's factor. The most intense collagen type IV staining was found to be in the region of the basement membrane and endothelial cells (Fig. 3).
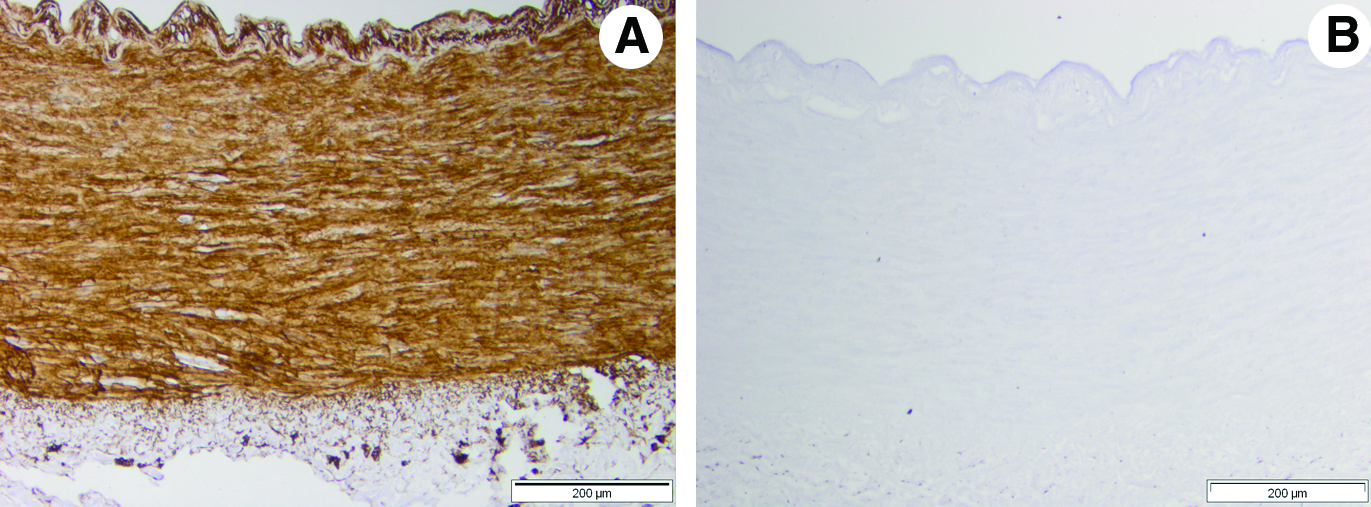
Representative histological images from the central region of fresh
Acellular arteries
Following decellularization, the arteries stained positively for collagen type I, fibronectin, laminin, and Von Willebrand's factor, with no obvious gross loss. Collagen type IV was notably absent following decellularization (Fig. 3).
Scanning electron microscopy
SEM examination focused primarily on the luminal surface of the arteries. The lumen of native common femoral arteries appeared to be lined with a continuous layer of endothelial-like cells exhibiting a typical elongated appearance (Fig. 4). There was a total absence of cells and cellular fragments on the luminal surface of acellular arteries (Fig. 4). The SEM images of acellular arteries demonstrated a smooth luminal surface. There was no evidence of damage to the basement membrane and exposure of the underlying collagen network.

Representative scanning electron micrographs of fresh
DNA content of acellular human arteries
Native and acellular common femoral arteries were processed using a DNA extraction kit. The extracted DNA was eluted from the spin columns and quantified spectrophotometrically, in which a peak of absorbance at 260/280 nm indicated the presence of DNA (the ratio of absorbance at A260 to A280 was greater than 1.4 in all cases). The data indicated that there was a greater than 97% (wet weight) reduction in DNA levels following decellularization (Table 1). Acellular arteries contained 0.007 μg·mg−1 of DNA whereas fresh arteries contained 0.273 μg·mg−1 (Table 1).
Data are expressed as the mean (n=6). The data for DNA content are expressed per mg, wet weight of tissue; the other data sets are expressed per mg of dry weight. The data presented demonstrate a significant lower DNA content in acellular arteries compared with native vessels. Further there were no significant differences between the collagen, GAGs, and denatured collagen content of acellular or fresh human arteries.
Biochemical assays
Hydroxyproline assay
There was no significant difference in the collagen content of acellular arteries compared with native tissue (ANOVA, p>0.05). The collagen content of native and acellular common femoral arteries was 700.1 and 686.7 μg·mg−1 respectively (Table 1).
Denatured collagen assay
There was no significant difference in the denatured collagen content of the native or acellular arteries (ANOVA, p>0.05). The denatured collagen content of native and acellular common femoral arteries was 12.5 and 10.1 μg·mg−1 respectively (Table 1).
Sulfated sugar assay
There was no significant difference in the sulfated sugar content of acellular arteries compared with native common femoral arteries. The GAG content of native common femoral arteries was 20.2 μg·mg−1 compared with acellular common femoral arteries, which was 17.7 μg·mg−1 (Table 1).
Contact cytotoxicity assay
Microscopic analysis of the contact cytotoxicity plates showed that 3T3 fibroblasts and baby hamster kidney cells grew up to and in contact with the samples of acellular common femoral arteries tested (Fig. 5). No zones of morphological change or cell lysis were noted (Fig. 5). Collagen alone (negative control) showed no signs of cytotoxicity (Fig. 5). Cyanoacrylate glue (positive control) caused cell lysis (Fig. 5).
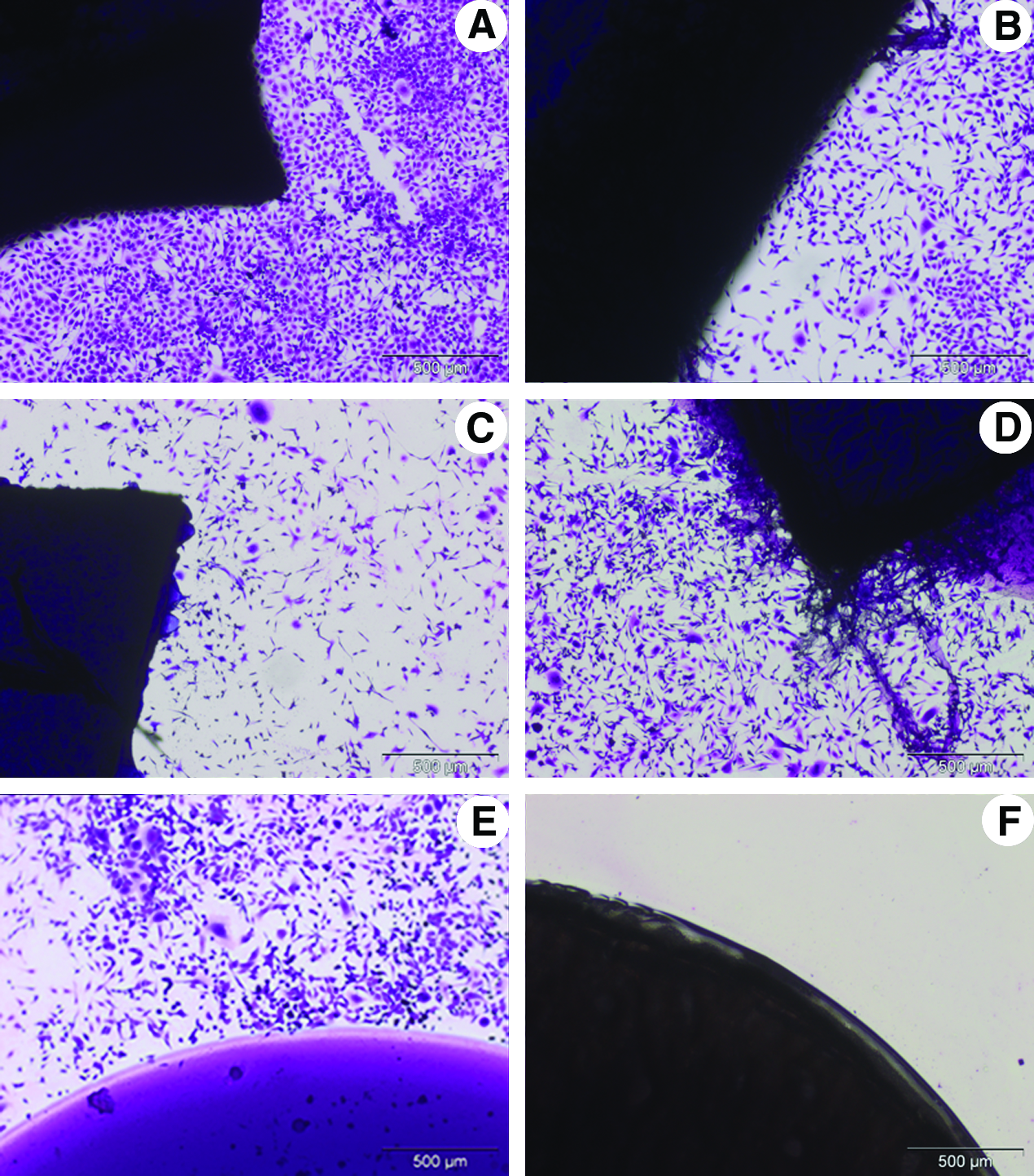
Contact cytotoxicity assessment of human common femoral arteries incubated with murine 3T3 fibroblasts
Extract cytotoxicity assay
There was no significant reduction in the cellular ATP levels of the 3T3 fibroblasts and baby hamster kidney cells following incubation with soluble extracts of acellular human common femoral arteries compared with that of the DMEM/GMEM negative control (ANOVA, p>0.05; Fig. 6). Extracts of fresh common femoral arteries were also screened using the same cell lines. The data demonstrated that there was a significant increase in ATP levels of baby hamster kidney cells cultured with extracts of fresh human common femoral arteries compared with GMEM (Fig. 6). There was, however, a significant reduction in the ATP levels of 3T3 fibroblasts cultured with extracts of fresh human common femoral arteries compared with DMEM (Fig. 6). The positive controls for cytotoxicity caused an almost total loss of ATP and were significantly lower than any other sample group (ANOVA, p<0.05; Fig. 6).
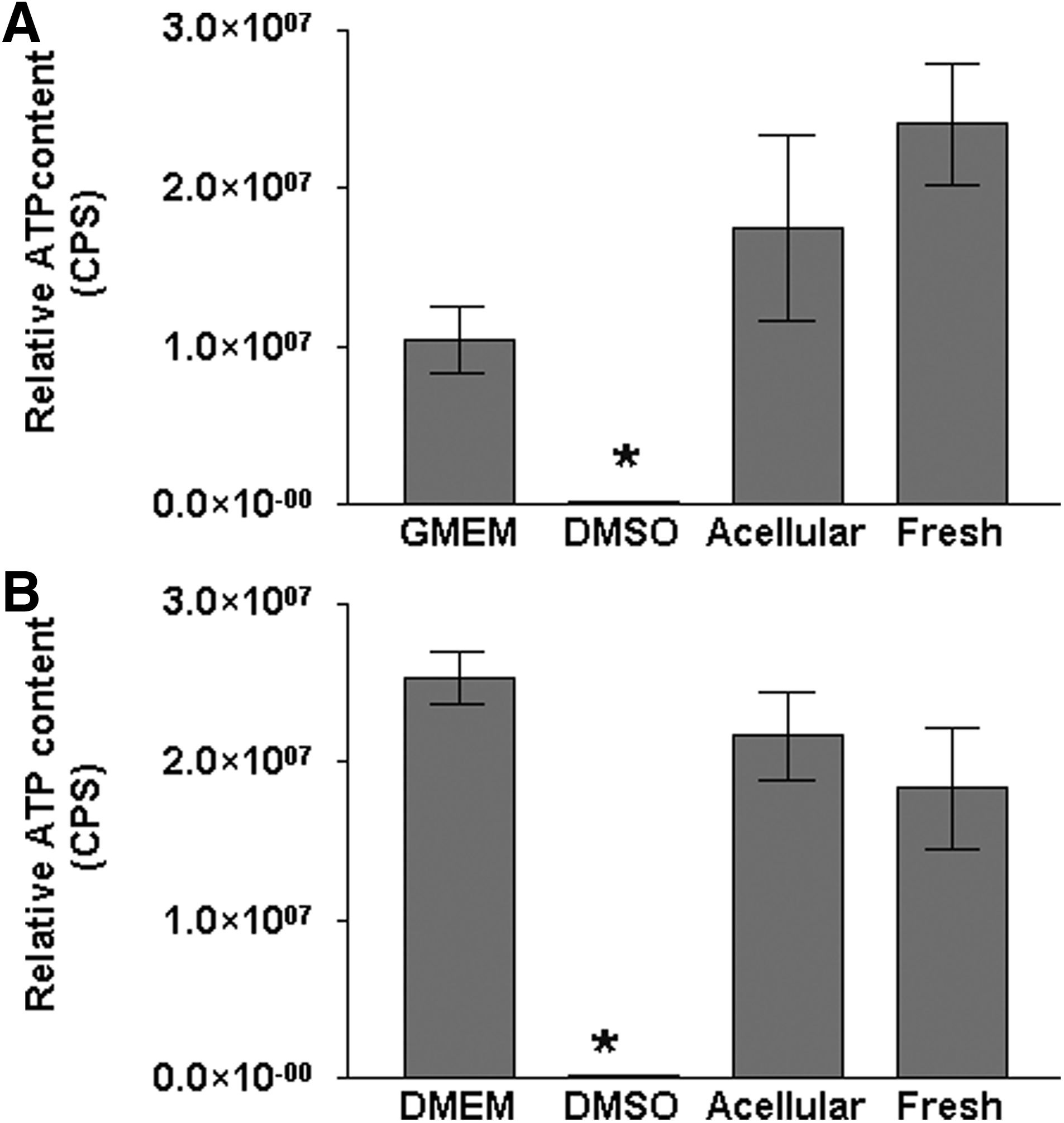
Extract cytotoxicity assay of fresh and acellular human common femoral arteries with in vitro–cultured murine 3T3 fibroblasts
Burst pressure testing
Acellular arteries were tested in a custom burst pressure rig and compared with native human arteries. The internal pressure was increased to a maximum of 3750 mmHg for each artery. The mean burst pressure of acellular common femoral arteries was 3214 mmHg compared with 2562 mmHg for native common femoral arteries. The data indicated that there was no significant difference between the burst pressures of the acellular arteries compared with native human arteries (ANOVA, p>0.05).
Tensile testing of native and acellular arteries
Following tensile testing of the native and acellular human arteries, it was found that there was no significant difference in elastin and collagen phase slopes, transition stress and strain, and ultimate tensile strength and failure strain in either the circumferential or axial direction (ANOVA, p>0.05; Table 2). All failures occurred at or near to the center of the gauge length.
Data are expressed as the mean (n=6)±95% confidence limits.
EI-E, elastic modulus; Coll-E, collagen modulus; UTS, ultimate tensile strength.
Suture retention testing
A single suture was pulled out of samples of native and acellular human common femoral arteries. The test was carried out using an Instron 5860 series table model testing systems at a speed of 10 mm·min−1. The data demonstrated that the maximum force in Newton's each artery was able to withstand before the suture was removed. There were no significant differences between maximum suture retention strength of native or acellular arteries (ANOVA, p>0.05; Fig. 7).
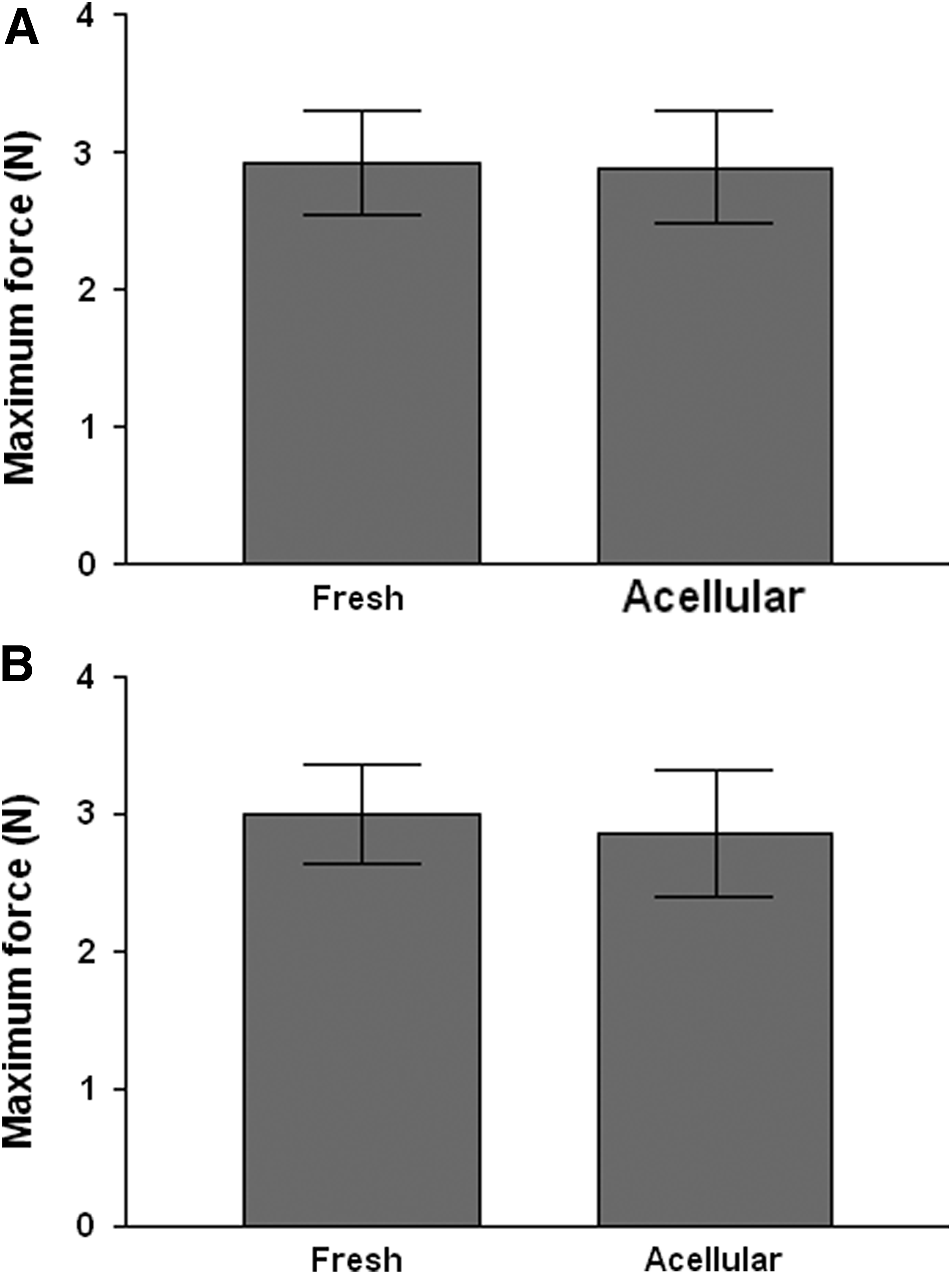
Suture retention strength of fresh and acellular human common femoral arteries in the
Compliance testing
The compliance testing indicated that there were no significant changes in the dilation of acellular human arteries compared with native controls. The dilation of native common femoral arteries at 200 mmHg was 7.9% compared with a dilation of 9.8% in acellular arteries.
Disinfection validation
Acellular common femoral arteries were subjected to treatment with and without peracetic acid. The ability of peracetic acid to penetrate the acellular matrices and the effectiveness of peracetic acid was determined against a defined microbial load of B. atrophaeus spores. Following incubation at 35°C, the number of colonies on each plate was recorded. The number of colonies counted was converted to CFU and a plot of total CFU against time was produced (Fig. 8). From this it was possible to determine the overall reduction in colony numbers and, therefore, log reduction at each time point (Fig. 8). Following treatment of acellular human common femoral arteries using 0% (v/v) peracetic acid, there was no apparent reduction in bacterial number over time. Approximately 80% of the bacteria were recovered at all time points (Fig. 8). Following 3 h incubation of acellular arteries with 0.1% (v/v) peracetic acid, a four-log reduction was shown and this was increased to a six-log reduction after 4 h, when no growth was detected (Fig. 8).
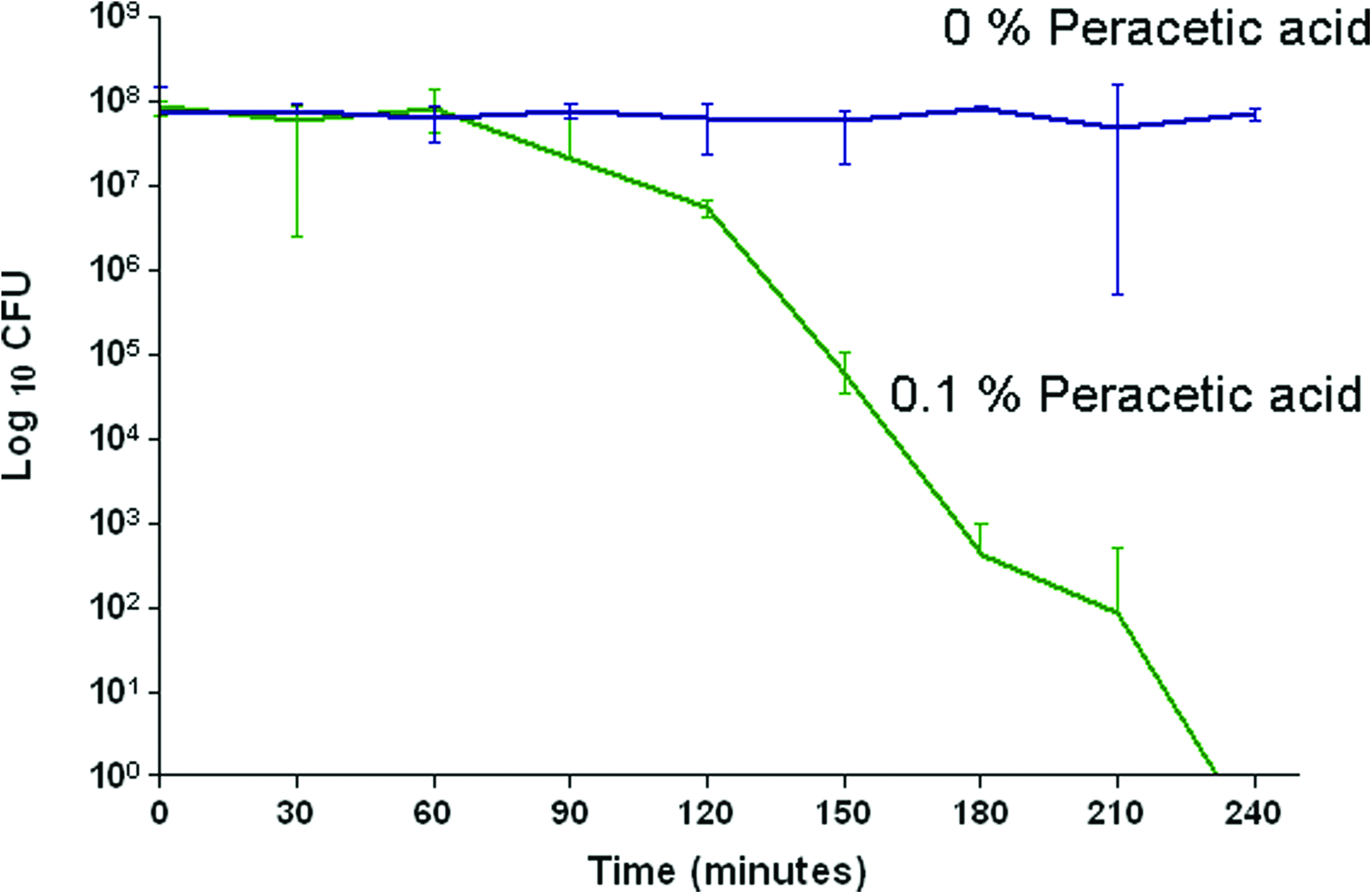
Change in bacterial load in response to treatment of acellular human common femoral arteries using 0% or 0.1% (v/v) peracetic acid. Data are expressed as mean (n=6)±95% confidence limits. The data supported the hypothesis that treatment with 0.1% (v/v) peracetic acid reduces the bioburden to undetectable levels of spiked acellular arteries following 4 h incubation at 27°C. Color images available online at www.liebertonline.com/tea
Discussion
The poor clinical performance of synthetic grafts composed of ePTFE or Dacron for small-diameter vascular applications has been the impetus for research toward biologically functional vascular grafts. Tissue engineering approaches have been at the forefront of this research, producing a variety of biological and synthetic substitutes; however, none have been successfully introduced into clinical practice. An ideal vascular graft should have similar biomechanics to the vessels being replaced or repaired, be resistant to thrombosis, and nonimmunogenic.
The use of ex vivo blood vessels as a scaffold for guided regeneration is becoming a common approach to the problem of small-diameter vascular grafts. However, the use of such vessels does necessitate a degree of processing to stabilize, sterilize, and prevent an adverse immune response. The host response to natural tissues is known to be largely dependent upon the method used to process the material. There are two popular approaches commonly utilized: cross-linking and removal of host cells and immunogenic epitopes using a decellularization protocol.
Chemical and nonchemical methods of cross-linking have been used extensively to modulate the physical, mechanical, or antigenic properties of naturally derived scaffolds.28–30 Glutaraldehyde cross-linking of tissues has been shown to reduce the immunogenicity together with providing the tissue with stability and mechanical durability; however, it also impairs some of the unique properties of the native tissue. The host response to cross-linked materials has been shown to be a foreign body type reaction with the formation of a fibrous capsule surrounding the implant. The cross-linking of collagen within the matrix prevents cellular infiltration and biodegradation of the matrix. 30 The inherent drawbacks of cross-linked materials are that they often retain cytotoxic compounds from the chemical processing, have the potential for calcification, and are incapable of cellular remodeling. As a result these cross-linked scaffolds are inert, behaving in a similar way to synthetic materials. The glutaraldehyde-fixed human umbilical vein graft has been shown to be an effective alternative to current synthetic grafts and offers improved patency rates. However, like other cross-linked materials, it is unable to remodel into a fully biological functional vessel.31,32
The aims of this study were to develop a process for the decellularization of human common femoral arteries, which had minimal effects on the tissue properties. Human common femoral arteries were treated using a novel decellularization process and were shown to be free from whole cells and cellular fragments. The acellular vessels retained the histioarchitecture and biomechanical properties of native arteries. Decellularization was achieved through modification of a protocol developed by Booth et al. 21 The decellularization protocol that was developed utilized low concentration of SDS and protease inhibitors. It was extremely mild and inhibited endogenous proteases released during cell lysis in an attempt to preserve the extracellular matrix. The first step of the process was a single freeze-thaw cycle to open up the arterial matrix making it more susceptible to the effects of the decellularization reagents. The arteries were treated using a disinfection solution to reduce the initial bioburden of the fresh tissue. A solution of 200 mM EDTA was used to detach endothelial cells from the luminal surface of the arteries. This was followed by sequential incubation in hypotonic tris buffer to lyse native cells, 0.1% (w/v) SDS in hypotonic buffer to solubilize cell fragments, and nuclease enzymes (RNAase and DNAase) to digest nucleic acids. The use of detergents and osmotic shock is a common approach taken to decellularize soft tissues.33–35 Hypertonic buffer followed by extensive washing in DPBS was used to remove any residual cellular remnants. Disinfection of the tissue was carried out using 0.1% (v/v) peracetic acid for 4 h. This approach has been used by a number of groups as a sterilant and to remove cells and cellular fragments from soft tissues.15,18 Residual peracetic acid was removed by extensive washing in DPBS.
Decellularization using SDS resulted in uniform cell removal throughout the arteries. This was confirmed by an absence of visible cells within the acellular arteries following staining using H&E. Histological analysis demonstrated that the acellular vessels had retained their native trilaminar arterial architecture. There was evidence of a dense medial layer and a thin adventitia layer, most of which was removed via blunt dissection prior to decellularization. Sirius red Miller's staining did not reveal any damage or loss of total collagen and elastin following decellularization. The images also confirmed retention of the internal elastic lamella, an important feature for endothelial cell attachment and resistance to thrombus formation. DAPI staining indicated an absence of whole cells and large DNA fragments from the acellular arteries. There was no histological evidence of loss of collagen type I, fibronectin, laminin, or Von Willebrand's factor from acellular arteries when compared with native samples. Immunohistochemical staining, however, identified a loss of collagen type IV from the basement membrane of acellular arteries. Collagen type IV is found exclusively in the basal lamina in the form of sheets and is believed to act as a scaffold for the binding and alignment of other basement membrane components such as laminin, entactin, and Heparan sulfate. Since collagen type IV forms into reticular structures, it maybe more susceptible to prolonged washing compared with other forms of collagen, which have been retained following decellularization. When other human tissues such as amniotic membrane or skin were treated using similar decellularization processes, collagen type IV immunostaining was unaffected.23,36 Collagen type IV and its associated components are known to play a role in cell adhesion and its loss may affect the potential for recellularization of the acellular matrix in vivo. However, the loss of collagen type IV immunostaining from the basement membrane did not have a detrimental effect on any of the biomechanical properties of the arteries. Whether the loss of collagen type IV has an effect on the vessel function in longer term will only be revealed by in vivo studies. The DNA content of acellular arteries was determined and acellular arteries retained around 3% of their original DNA content. DNA reduction is an important outcome of decellularization, as it is indicative of cell number. Moreover, any large DNA fragments within the extracellular matrix have the potential to act as a nucleation site for calcification. A lack of substantial DNA within a therapeutic human tissue graft is highly a desirable feature. The in vitro biocompatibility of acellular human arteries was demonstrated using extract and contact cytotoxicity assays. B. atrophaeus spores were used to determine the validity of using peracetic acid as a chemical disinfectant within the decellularization process. Biochemical analysis demonstrated that there were no differences in the composition of the extracellular matrix of acellular arteries when compared with native samples. There was no loss of collagen from the matrix and conversely there was no increase in the denatured collagen content of the arteries as a result of decellularization. Burst pressure and compliance are the arguable key factors in determining the suitability of any vascular graft. There was no significant difference in the burst pressure of acellular human arteries when compared with native controls. The burst pressure of acellular arteries far exceeded normal arterial pressures likely to occur in vivo at around 3214 mmHg. These values were considerably higher than those of fresh and SDS-treated human saphenous veins, which were reported to be 2480 and 2380 mmHg respectively. 18 Studies using automated dissection and decellularization using 1% (w/v) SDS and 75% (v/v) ethanol resulted in veins with much lower burst pressures, 972 mmHg. 19 The burst pressure of a tissue-engineered blood vessel produced using multiple sheets of human fibroblast was 3040 mmHg; these were, however, in culture for ∼20 weeks. 10 When subjected to increasing internal pressures, fresh and acellular arteries exhibited a similar dilation profile. The compliance of decellularized arteries has not been widely reported in the literature. The compliance of human internal mammary arteries has been reported to be 11.5%, compared with a value of 3.4% for a blood vessel produced using multiple sheets of human fibroblasts. The tissue-engineered vessel was stiffer than fresh internal mammary arteries; however, following 6 months after implantation, the vessel became more elastic with a compliance of 8.8% at 100 mmHg. These values were found to be similar to those reported in the literature for the dilation of human carotid arteries. 37 Matching the biomechanical properties of vascular grafts to the implantation site should prevent a compliance mismatch and reduce the potential for thrombus formation and failure of the graft. Low strain rate failure testing indicated that there was no significant difference in elastin and collagen phase slopes, transition stress and strain, and ultimate tensile strength and failure strain when acellular arteries were compared with native vessels. Suture holding capacity is an important property, which may demonstrate the utility of any surgical material. The suture pullout values of acellular allogeneic common femoral arteries were not significantly different from native controls. The suture holding capacity for human umbilical vein was determined to be 1.68 N compared with 2.21 N for decellularized vein. 19 These values were significantly lower than those obtained for fresh and acellular human arteries (∼3 N), which was expected owing to the superior biomechanical characteristics of artery.
Decellularization has been successfully applied to a wide variety of human and xenogeneic tissues such as aortic cardiac valves, pulmonary roots, pericardium, amniotic membrane, skin, and many others.23,38–41 Indeed, acellular allogeneic tissues have been used clinically in cardiovascular applications. 42 Small intestinal submucosa is the most characterized and widely clinically used decellularized xenogeneic product. Small intestinal submucosa (SIS) has been used without major complication for a number of years in applications ranging from hernia repair and fistula closures. 14 However, decellularized tissues have not always been as successful.43,44 Decellularization is a finely balanced process of controlled damage to tissues to remove cells and nucleic acids while observing the tissue architecture. The main advantages of decellularized grafts are their inherent biomechanical properties; however, it is vital to match these as closely as possible to the site of implantation. Compliance mismatch can ultimately influence the clinical outcome of an implanted artery. 9
Decellularization of human arteries has the potential to offer many benefits over currently available preserved human arterial grafts. Cryopreserved human grafts have been shown to contain viable cells, and while this results in good patency rates, the allogeneic cells have the potential to evoke an immune response. Allogeneic cells within the graft will be recognized as foreign by the recipient and maybe rejected depending upon how closely the donor and recipient are matched genetically. Moreover acellular arteries will not require specialist storage facilities and this has the potential to make the grafts universally accessible to clinicians.
The data presented indicate that an acellular human artery has the potential for development of a small-diameter vascular graft for bypass surgery or hemodialysis access. The results demonstrate that the decellularization procedure was successful in removing cells from the native arteries along with >97% of total DNA. The acellular arteries were shown to be biocompatible in vitro. This was achieved while preserving the major biochemical components of the vessels and not significantly altering the biomechanical properties. However, the acellular arteries will need to be evaluated in a functional animal model to determine any immune reaction and patency prior to clinical use.
Footnotes
Acknowledgments
Funding support was from the Department of Health [HTD 430], from the NHS BT TS, and from the Tissue Regenix Group plc.
This work was partially funded through WELMEC, a Centre of Excellence in Medical Engineering funded by the Wellcome Trust and EPSRC, under grant number WT 088908/Z/09/Z.
Disclosure Statement
Professors E. Ingham and J. Fisher are Directors of Tissue Regenix Group plc, to which the decellularization technology is licensed. Professor S. Homer-Vannaisinkam is Chair of the clinical advisory board for Tissue Regenix Group plc.