
Abstract
There is a need to improve the quality of donor liver from donation after circulatory death (DCD). The purpose of this study was to investigate the effects and mechanism of normothermic machine perfusion (NMP) combined with bone marrow mesenchymal stem cells (BMMSCs) on the oxidative stress and mitochondrial function in DCD livers. DCD livers were obtained, a rat NMP system was established, and BMMSCs were extracted and identified. The DCD livers were grouped by their preservation method: Normal, static cold storage (SCS), NMP (P), and NMP combined with BMMSCs (PB), and the preservation time was up to 8 h. An IAR20 cell oxidative stress injury model was established in vitro by simulating DCD oxidative stress injury and coculturing with BMMSCs for 6 h. Compared with SCS group, after 6 h in vitro, the PB and P groups had significantly improved liver function and liver histological damage, reduced hepatocyte apoptosis and oxidative stress, improved hepatocyte mitochondrial damage, and increased mitochondrial membrane potential. These indicators were significantly better in the PB group than in the P group. BMMSCs significantly inhibited reactive oxygen species release from the IAR20 cell oxidative stress model in vitro, ameliorated mitochondrial damage, and increased mitochondrial membrane potential level. BMMSCs also downregulated the JUN N-terminal kinase-nuclear factor kappa B (JNK-NF-κB) signaling pathway significantly in the IAR20 cell oxidative stress model and promoted AMP-activated protein kinase (AMPK) activation. We verified that NMP combined with BMMSCs also played the same role in the PB group. NMP combined with BMMSCs could improve liver quality by relieving oxidative stress injury and improving mitochondrial function in rat DCD livers. The mechanism of protective role might involve inhibiting the JNK-NF-κB pathway to reduce oxidative stress and promote AMPK activation, thereby reducing mitochondrial damage and increase mitochondrial function.
Introduction
With the emergence of liver donor shortage, obtaining an effective donor liver is an important issue for liver transplantation [1]. The emergence of donation after circulatory death (DCD) livers provides new ideas to solve the problem of donor liver shortage. However, DCD livers experience a long period of warm ischemic injury and ischemia–reperfusion injury (IRI), which makes them prone to complications and rejection after surgery, and seriously affects the survival of donor livers and patient prognosis [2 –4]. Thus, the use of DCD livers has certain risks and limitations; therefore, improving the quality of DCD livers is an important and urgent research field [4].
Static cold storage (SCS) has a limited ability to preserve marginal donor liver, because DCD livers experience cold ischemia, do not have sufficient physiological reserves to tolerate SCS-related injuries, and liver energy metabolism and mitochondrial function are easily damaged, making them particularly sensitive to IRI; therefore, we should limit their exposure to SCS [5,6]. Normothermic machine perfusion (NMP) mimics the normal metabolic state in vivo and can preserve and repair donor livers, providing a strong advantage in the preservation of high-risk livers and marginal donors, and can more effectively increase the use of donor livers, thus NMP is very promising to expand the donor pool [7 –9].
However, although a large number of studies have confirmed that the in vitro NMP system can, to some extent, repair DCD livers through the supply of energy and nutrients, research also found that although preservation by NMP alone can provide energy and nutrients, it cannot completely avoid bile duct damage [10] and oxidative stress injury caused by ischemia-reperfusion [11], nor control the activation of Kupffer cells and endothelial cells [12]; therefore, it is necessary to find a new method based on NMP that can more comprehensively repair DCD livers.
Bone marrow mesenchymal stem cells (BMMSCs) are a class of nonhematopoietic stem cells derived from stromal cells that can reduce hepatocyte damage and accelerate liver regeneration, participating in anti-inflammatory pathways and regulating immunity [13 –18]. BMMSCs have marked targeted reparative effects on damaged organs in multiorgan applications, and have protective and reparative effects in kidney transplantation and lung injury applications [19 –21]. Previous studies have confirmed that BMMSC transplantation can reduce liver IRI and inhibit postoperative hepatocyte apoptosis [22]. Therefore, the present study aimed to explore the reparative effect and molecular mechanism of NMP combined with BMMSCs on DCD livers.
Materials and Methods
Animals and materials
We purchased specific pathogen-free rats from the Food and Drug Administration (Beijing, China). The animals were kept for 2 weeks at 50% humidity, 18°C–23°C, and under 12 h light–dark, with ad libitum access to food and water. We replaced the cages and bedding regularly. All animals received humane care in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th edition). The Animal Care and Research Committee of Tianjin First Central Hospital (Tianjin, China) approved all the protocols (Permit No.: 2016-03-A1). BMMSCs were extracted from healthy male Sprague-Dawley (SD) rats (n = 15, 4–5 weeks old, 40–60 g); livers were obtained from healthy male SD rats (n = 50, 6–8 weeks old, 200–220 g).
The following reagents and kits were used in the present study: Dulbecco's modified Eagle's medium (DMEM)/F12 medium (1:1); 0.25% trypsin–EDTA solution (Gibco, Carlsbad, CA, USA); penicillin–streptomycin solution (HyClone, Logan, UT, USA); fetal bovine serum (FBS; Biowest, Loire Valley, France); adipogenic and osteogenic differentiation medium (Sigma-Aldrich, Merck KGaA, St. Louis, MO, USA); Oil Red O (Beijing Dingguo Changsheng Biotechnology, Beijing, China); a von Kossa cell staining kit (Genmed, Shanghai, China); rat green fluorescent protein genomic adenovirus (GFP-Adv, GeneChem, Shanghai, China); proteinase K and a bicinchoninic acid (BCA) protein assay kit (Beijing Solarbio Science & Technology, Beijing, China); western blotting-related reagents, a reactive oxygen species (ROS) assay kit, a tissue mitochondrial isolation kit, and a mitochondrial membrane potential assay kit with JC-1 (Beyotime, Shanghai, China); anti-rat CD34-fluorescein isothiocyanate (FITC), anti-rat CD29-phycoerythrin (PE), anti-rat CD45-PE, anti-rat CD90-FITC, anti-rat RT1 class I gene (A-1) (RT1A)-PE, anti-rat RT1 class II, locus B (RT1B)-FITC (BioLegend, San Diego, CA, USA); JUN N-terminal Kinase (JNK) mouse antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA); phosphorylated (p)-nuclear factor kappa B (NF-κB) (p-p65) rabbit antibody, p-JNK rabbit antibody, p-AMP-activated protein kinase (AMPK) rabbit antibody, p-acetyl-CoA carboxylase (ACC) rabbit antibody, ACC rabbit antibody (Cell Signaling Technology, Boston, MA, USA); NF-κB (p65) rabbit antibody, AMPKα rabbit antibody, myeloperoxidase (MPO) (Proteintech, Wuhan, China); β-actin mouse antibody, goat anti-rabbit immunoglobulin G-horseradish peroxidase (IgG-HRP), goat anti-mouse IgG-HRP, FITC-conjugated goat anti-rabbit IgG, streptavidin–peroxidase kit (ZSGB-BIO, Beijing, China); malondialdehyde (MDA) and glutathione (GSH) assay kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China); a annexin V-FITC/PI apoptosis detection kit (Keygen Biotech, Nanjing, China); inverted fluorescent microscope (Olympus, Tokyo, Japan); Eclipse Ni-U-positive fluorescence microscope (Nikon); BD Accuri C6 Plus flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA); Molecular Imager ChemiDoc XRS+ system (Bio-Rad, Foster City, CA, USA); an in vivo imaging system (PerkinElmer, Fremont, CA, USA); and a transmission electron microscope (Hitachi, Ltd., Tokyo, Japan).
BMMSC isolation, culture, and identification
Rats were sacrificed by cervical dislocation, and the femur and tibia were removed aseptically. The marrow cavity was rinsed with DMEM/F12 (1:1) containing 10% FBS; the cell suspension was inoculated into T75 culture flasks, and cultured at 37°C with 5% CO2. Flow cytometry identification: passage 3 cells were labeled with fluorescent antibodies (anti-CD29-PE, anti-CD34-FITC, anti-CD45-PE, anti-CD90-FITC, anti-RT1A-PE, and anti-RT1B-FITC) and incubated for 30 min in the dark for flow cytometry. The identification of in vitro adipogenic and osteogenic differentiation of BMMSCs was performed using the method described by Yang et al. [23].
BMMSC colonization in the liver
To generate GFP-BMMSCs, the spent medium was removed from passage 3 BMMSCs and replaced with 5 mL DMEM/F12 per flask. GFP-Adv transduction solution was added at a multiplicity of infection of 10. After 6 h, complete medium was added, and the culture medium was changed every other day. After 72 h, the proportion of GFP-expressing cells was observed using fluorescence microscopy. BMMSC colonization was detected in frozen liver sections from GFP-BMMSC plus 6-h NMP (protected from light during perfusion) livers. At the end of the 6-h NMP incubation, about 1 × 1 × 0.5 cm3 liver tissue blocks were randomly removed and embedded in OCT glue, and quick-frozen in liquid nitrogen. Sections (10-μm thick) were obtained, fixed in 4% paraformaldehyde, and observed using fluorescence microscopy. GFP-BMMSCs tracer detection: after perfusion, the entire liver was removed and the colonization of GFP-BMMSCs in the liver was observed using a live imaging system.
Rat DCD liver acquisition and establishment of the liver NMP system
The rats were fasted for 12 h, but allowed access to water. The rats were anesthetized by intraperitoneal injection of 5% chloral hydrate (10 mL/kg). The liver was exposed using an abdominal median incision. The hepatic artery was ligated and the portal vein was separated. The choledochus was intubated and the bile drained. After the diaphragm was opened, the thoracic aorta was clipped, and the heart was pressed with a cotton swab to simulate cardiac death. The abdominal cavity was covered with gauze soaked in warm saline for 30 min, and then the liver was harvested and weighed to determine its wet weight. For the detailed method, please refer to the method described by Yu et al. [24].
The NMP system is a single-cycle system that includes centrifugal pumps, membrane oxygenators, organ chambers, heaters, and pressure and temperature monitors. The NMP system was maintained at 35°C–38°C. The perfusate ingredients were 60 mL DMEM/F12 (1:1) containing 20% FBS and 1% penicillin–streptomycin solution (penicillin 10,000 U/mL, streptomycin 10,000 μg/mL), 20 mL of fresh blood, 5 U/mL of heparin, 2 U/L of insulin, and 2.5 μg/mL of dexamethasone. The DCD liver was placed in the organ chamber and the perfusion system was connected in advance. The perfusate was oxygenated through a membrane oxygenator and flowed through the portal vein, and was continuously perfused at a rate of 2 mL/g/min (by liver wet weight). The portal pressure was maintained at 10–12 mm H2O and monitored using a pressure sensor (Fig. 1A, B).
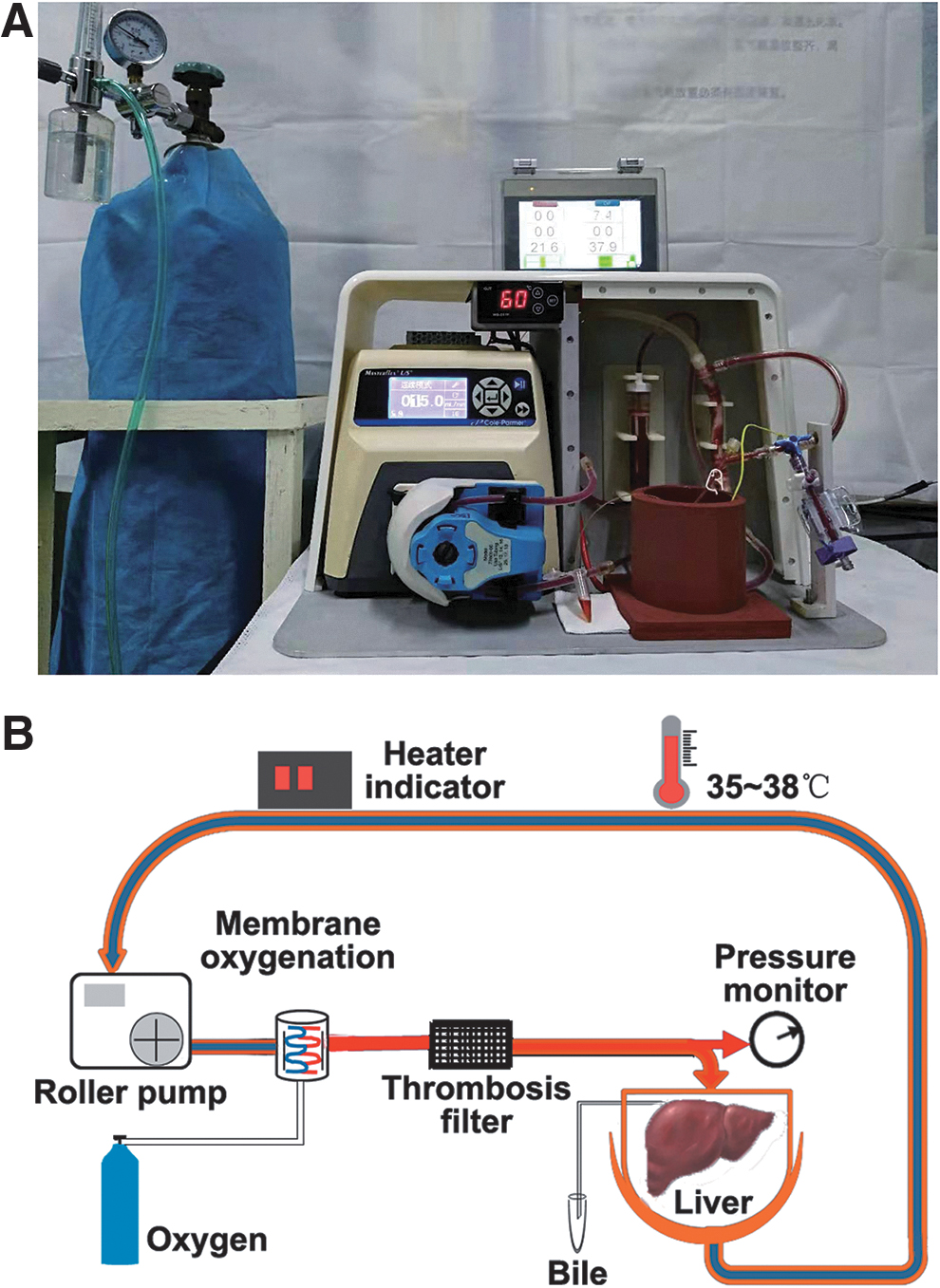
Rat NMP system.
Groups and treatments for DCD livers
Livers were grouped into normal, SCS, NMP alone (P), and BMMSCs plus NMP (BP) groups. In the Normal group, serum and livers were obtained for use. The blood was washed from the SCS livers using 20 mL of University of Wisconsin solution (UW) at 4°C and then the livers stored at 4°C in UW. The livers were harvested after 4, 6, and 8 h. In the NMP group, 2 mL of normal saline was injected via the portal vein immediately after the NMP system was connected, and NMP was performed continuously. In the BP group, 2 mL of medium suspension containing 1–3 × 107 BMMSCs was injected via the portal vein immediately after the NMP system was connected. Five DCD livers per group were used at each time point. The inflow perfusate was collected for blood gas analysis at the instant of perfusion and at 2, 4, 6, and 8 h. The inferior vena cava outflow perfusate was centrifuged to obtain the supernatant. Liver specimens were collected at the instant of perfusion, and 2, 4, 6, and 8 h. Liver tissues were randomly fixed in formalin or 2.5% glutaraldehyde solution, and stored at 4°C; other liver tissues were minced and quick-frozen with liquid nitrogen. Partial samples were stored at −80°C before testing.
In vitro model of oxidative stress of hepatocytes
Establishment of an oxidative stress injury model of IAR20 cells in vitro
Rat liver parenchymal cell line IAR20 was cultured with minimal essential medium/Earle's balanced salt solution containing 20% FBS and 1% penicillin–streptomycin solution at 37°C in a 5% CO2. IAR20 cells were seeded in a six-well plate with a cell density of 5 × 105/well. After 24 h, the cells were stimulated with H2O2 at concentrations of 0, 0.1, 0.2, 0.4, and 0.6 mmol/L for 30 min. The medium was changed and the cells were cultured for 6 h. IAR20 cells and supernatant were collected for subsequent detection.
IAR20 cells cocultured with BMMSCs after oxidative stress
IAR20 cells were inoculated into six-well plates at a cell density of 2 × 105/well, and H2O2 was added at a concentration of 0.2 mmol/L for 30 min. After the medium was changed, different experimental treatments were performed. The groups comprised IAR20 (I) group, IAR20 (H2O2) (IH) group, and IAR20 (H2O2) cocultured with BMMSCs (IH B) group. BMMSCs were inoculated in the Transwell chamber at a cell density of 106/well, the cells were cultured for 6 h. IAR20 cells and supernatant were collected for subsequent detection.
Liver function
Samples (200 μL) of inferior vena cava outflow perfusate supernatant or cell supernatant were taken to detect alanine aminotransferase (ALT), aspartate aminotransferase (AST), and mitochondrial AST (ASTm).
Oxidative stress injury detection
For the MDA test, liver tissue and IAR20 cells were collected to detect the degree of lipid peroxidation. For the GSH test, liver tissue and IAR20 cells were collected to detect the degree of peroxidative damage and antioxidant capacity. The assays were carried out following the instructions of the assay kit.
For IAR20 cell viability test, IAR20 cells were inoculated into 96-well plates at a cell density of 1 × 104/well, and 24 h later, they were stimulated with H2O2 at concentrations of 0, 0.1, 0.2, 0.4, and 0.6 mmol/L for 30 min. The medium was changed, the CCK-8 reagent was added, and incubation was continued for 6 h before the cell viability was detected.
To test the cellular ROS levels, the 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescent probe method was used. IAR20 cells were suspended in diluted DCFH-DA, and incubated for 20 min. Cells were gently shaken every 3–5 min to fully contact the cells with the probe. After incubation, the cells were washed three times with FBS-free medium to remove the DCFH-DA that did not enter the cells, and the cells were detected using flow cytometry.
Liver histopathological and immunofluorescence staining
For hematoxylin and eosin (HE) staining, liver tissue was fixed, paraffin-embedded, sectioned, and stained with HE. Hepatic pathological changes were observed using light microscopy. Hepatic IRI severity was evaluated based on Suzuki's criteria [25]. For paraffin section immunofluorescence (IF), tissue slides underwent deparaffinization with dimethylbenzene, gradient ethanol hydration, antigen retrieval, and blocking with normal goat serum. The slides were incubated with primary antibodies (1:50) at 4°C overnight, with secondary antibodies comprising FITC-conjugated goat anti-rabbit IgG at room temperature for 2 h, the nuclei were stained with 4′ 6-diamidino-2-phenylindole (DAPI), and the expression of MPO in the liver was observed under a fluorescence microscope. For cell IF, cell slides were washed three times with phosphate-buffered saline, fixed with 4% paraformaldehyde for 20 min, permeated with 0.5% Triton X-100 for 20 min at room temperature, and blocked with normal goat serum for 30 min at room temperature. The slides were incubated with primary antibodies (1:50) at 4°C overnight, with secondary antibodies comprising of FITC-conjugated goat anti-rabbit IgG at room temperature for 1 h, and the nuclei were stained with DAPI, and the expression of MPO in IAR20 cells was observed under a fluorescence microscope.
Apoptosis detection
Liver tissue slides were obtained, deparaffinized with dimethylbenzene, hydrated in gradient ethanol, and permeabilized using 10 μg/mL proteinase K at 37°C for 20 min. The reaction solution (labeling solution:enzyme solution = 50:1) was added and the slides were incubated at 37°C for 1 h. The nuclei were stained with DAPI, and hepatocyte apoptosis was observed under a fluorescence microscope.
IAR20 cell apoptosis was detected using Annexin V-FITC/PI double staining. Cells were collected and 5 μL each of Annexin V-FITC and PI were added and the cells were incubated for 15 min (protected from light) at room temperature; flow cytometry detection was then performed.
Mitochondrial morphology and membrane potential detection
Fresh liver tissue was cut into 1 × 1 × 2 mm3 samples, and IAR20 cell pellets were collected. Samples were fixed in 2.5% glutaraldehyde solution, embedded, and sliced into ultrathin sections. The ultrastructural changes were observed under a transmission electron microscope.
For mitochondrial membrane potential detection, fresh liver tissue mitochondria were collected and loaded with JC-1 fluorescent probes to detect mitochondrial JC-1 aggregates and monomers using a fluorescence microplate reader. IAR20 cells were loaded with JC-1 fluorescent probes, and mitochondrial JC-1 aggregates and monomers were detected using microscopy and flow cytometry. The ratio of mitochondrial JC-1 aggregates and monomers was compared to evaluate the mitochondrial membrane potential level, following the instructions of the assay kit.
Western blotting
Total proteins from livers or IAR20 cells were extracted using Radioimmunoprecipitation assay lysis buffer; the total protein concentration was quantified using the BCA method. The proteins were separated electrophoretically and wet-transferred to polyvinylidene fluoride (PVDF) membranes, blocked for 1 h with 5% skimmed milk (BD Biosciences), and then incubated with primary antibodies against JNK, p-JNK (Thr183/Tyr185), NF-κB (p65), p-NF-κB (Ser36), AMPKα, p-AMPK (Thr172), ACC, p-ACC (Ser79) (1:500), and β-actin (1:2,000) at 4°C overnight. The membranes were rinsed with Tris NaCl with Tween20 buffer, and incubated with secondary antibodies (1:2,000) at room temperature for 1 h. The chemiluminescence HRP substrate was added to the PVDF membrane, the membranes were exposed, and image grayscale values were analyzed using AlphaView SA 3.4.0.0 (ProteinSimple, San Jose, CA, USA).
Statistical analysis
The data were analyzed using SPSS 17.0 (IBM Corp., Armonk, NY, USA). The mean ± the standard deviation was used to present normally distributed data. One-way analysis of variance was used to assess the significance of differences between groups; least significant difference and Student–Newman–Keuls post hoc comparisons were used for further analyses. Statistically significant differences were indicated using P < 0.05. Data were plotted for presentation using GraphPad Prism 5.0 (GraphPad, La Jolla, CA, USA).
Results
BMMSC morphology, identification, and colonization in the liver
The BMMSCs were long and spindle-shaped, and appeared partially vortexed or chrysanthemum-like, with typical mesenchymal stem cell (MSC) morphological characteristics. Flow cytometry showed that the proportions of CD29+CD34− cells, CD90+CD45− cells, and RT1A+RT1B− cells were 100.0%, 99.9%, and 99.7%, respectively, indicating that the passage 3 BMMSCs were highly pure. Oil Red O staining showed several red lipid droplets in the cytoplasm after adipogenic induction, which was consistent with adipocyte characteristics. Von Kossa staining showed black granular or lumpy calcium deposits in the cytoplasm after osteogenic induction, which is an osteoblast characteristic. The results indicated that the extracted BMMSCs could differentiate into adipocytes and osteoblasts (Fig. 2A–F).
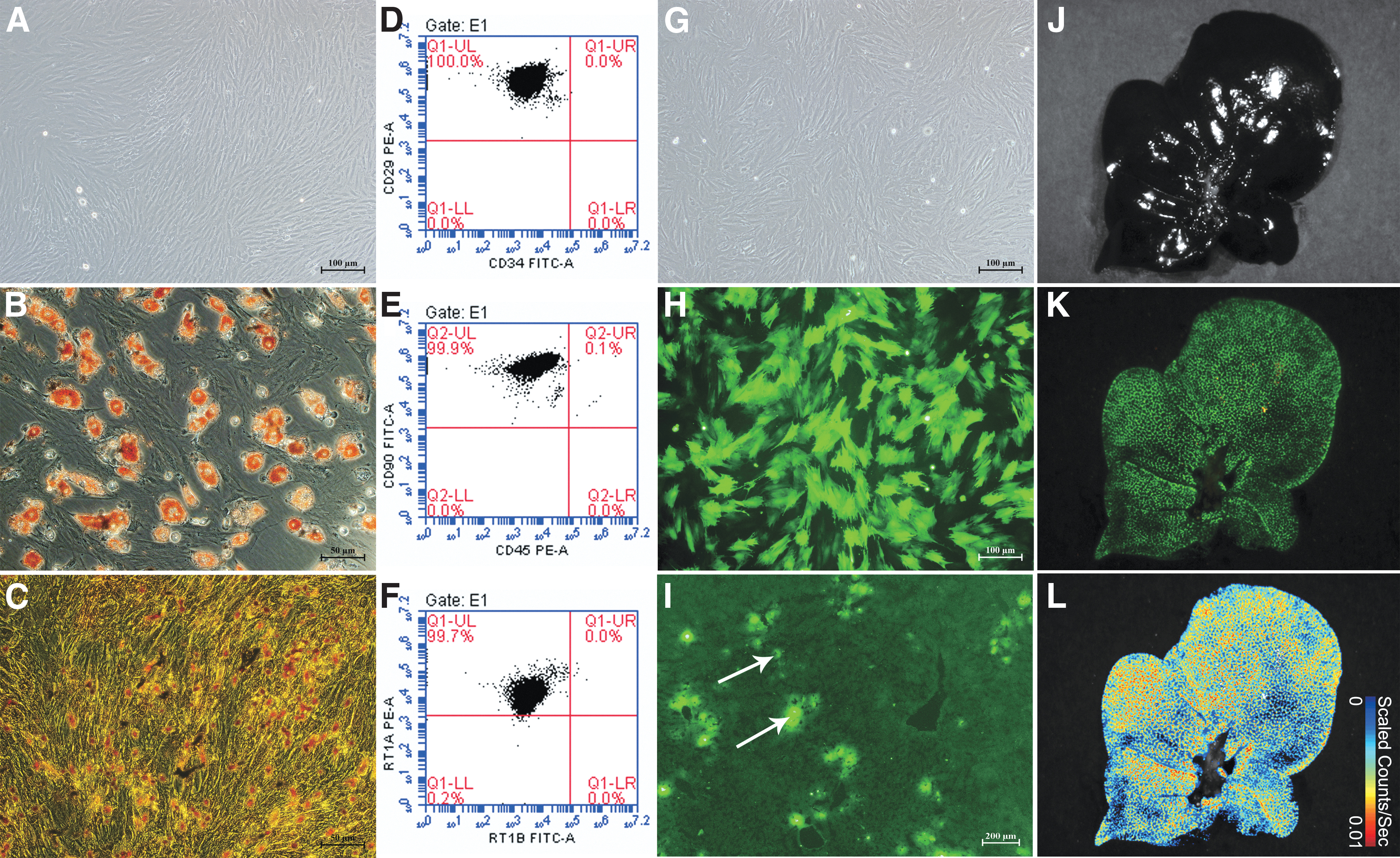
BMMSC morphology, identification, and colonization in the liver.
After GFP-Adv transduction, more than 85% of the of BMMSCs expressed GFP, which proved the successful transfer of the GFP gene into the BMMSCs, and the successful construction of GFP-BMMSCs (Fig. 2G–I). After 6 h of perfusion, GFP-BMMSCs (green fluorescence) had colonized the hepatic sinusoids, as detected in frozen liver sections. Tracer detection showed that most BMMSCs continuously colonized the liver. The results suggested that during DCD liver repair, BMMSCs could colonize in the hepatic sinusoids (Fig. 2J–L).
Establishment and quality evaluation of the rat NMP system
ALT and AST levels showed an increasing trend, which slowed gradually, and then decreased significantly from hour 4 to 6. The ALT and AST levels were not significantly different from hour 4 to 6, but were significantly different between the other time points. After 6 h of perfusion, ALT and AST levels were significantly elevated (P < 0.05). Lactate rapidly decreased to low levels after perfusion for 6 h. After 6 h of perfusion, there was an evident increase in lactate; the difference between the levels at hour 2 and 6 was significant (P < 0.05). Bile increased gradually, but decreased after 6 h of perfusion (Fig. 3A).
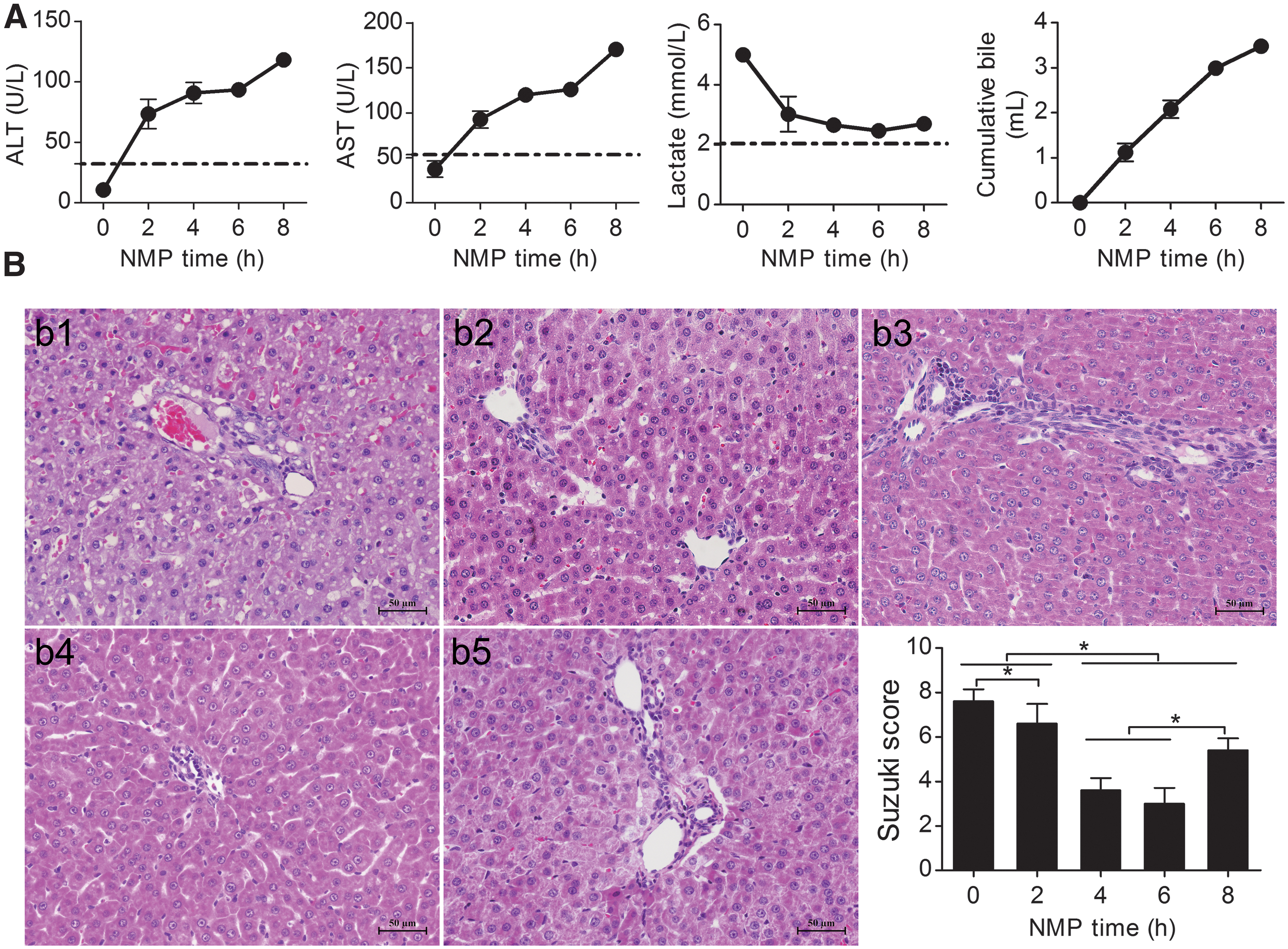
Rat NMP system evaluation.
Liver histopathology showed severe cytoplasmic vacuolization immediately after perfusion; severe hepatic sinusoid congestion, cell edema, and acidophilic degeneration were evident. The cytoplasmic vacuolization degeneration, cell edema, and hepatic sinusoid congestion gradually decreased along with perfusion time. At hour 6, there was no obvious cell edema, vacuolization degeneration, acidophilic degeneration, or hepatic sinusoid congestion, and the liver histopathology was the best at hour 6. Hepatocyte edema, acidophilic degeneration, and necrosis were observed at hour 8. The Suzuki scores were significantly different at each time point, and were lower at hour 4 and 6, but were not significantly different. The Suzuki score increased significantly at hour 8, suggesting that IRI improved gradually with perfusion time; however, the liver appeared injured after 6 h of perfusion (Fig. 3B).
Evaluation of liver function, lactate clearance, bile production, and histopathology in the rat NMP system suggested that this preservation method could significantly improve DCD liver function and histopathology. The DCD liver quality improved gradually with perfusion time; however, liver function, lactate clearance, bile production, and histopathology deteriorated after 6 h of perfusion, and the DCD liver quality decreased, suggesting that the best and longest time for storing DCD livers in the rat NMP system was 6 h and that further perfusion might negatively affect DCD liver quality.
NMP combined with BMMSCs improved DCD liver quality
NMP combined with BMMSCs improved DCD liver function
ALT and AST levels showed an increasing trend, which slowed gradually with perfusion time. The PB group had significantly lower ALT and AST levels than the P group (P < 0.05). Liver function tests suggested that NMP combined with BMMSCs could improve DCD liver function and quality significantly, and was superior to NMP alone (Fig. 4A).
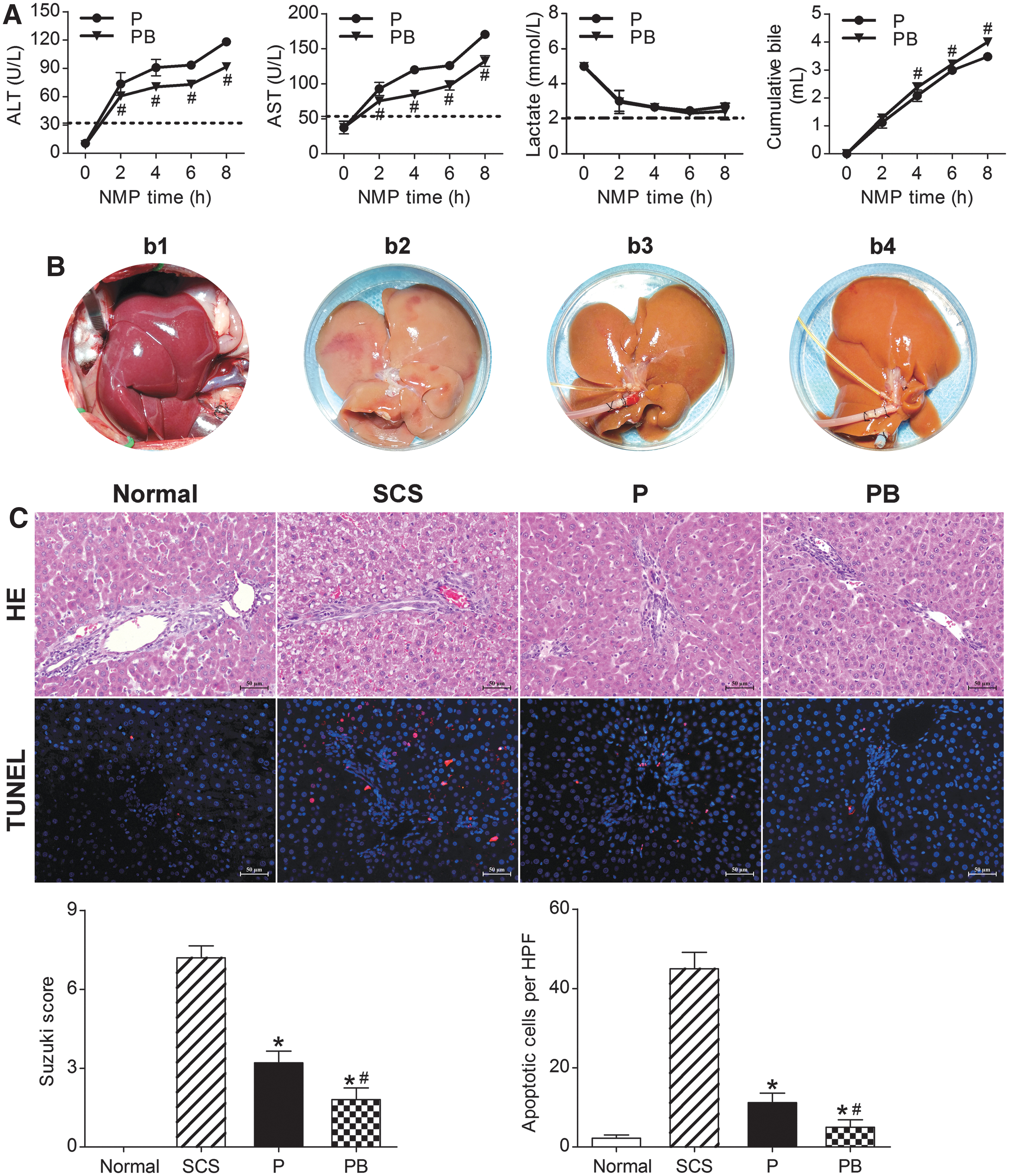
NMP combined with BMMSCs improved DCD liver quality.
Lactate levels were highest immediately after perfusion, and decreased gradually to stable levels with increasing perfusion time. After 6 h of perfusion, there was an evident increase in lactate; the PB group had lower lactate levels than the P group at each time point. Bile gradually increased with perfusion time. The PB group had significantly higher bile production and rate of increase than the P group at each time point (P < 0.05). After 6 h of perfusion, the P group had a significantly slower bile increase rate, suggesting that NMP combined with BMMSCs significantly improved DCD liver quality and was superior to NMP alone (Fig. 4A).
NMP combined with BMMSCs improved DCD liver general performance and histopathology
After 30 min of warm ischemia, the DCD livers were obviously swollen, with severe congestion, purple-red color, and rounded edges. After 6 h of SCS, the livers were swollen, with an uneven texture, congested, and piebald-like. The P and PB livers were not evidently swollen or congested, the texture was uniform, and with a soil yellow color, suggesting that NMP alone and combined with BMMSCs could reduce liver swelling and congestion, and improve performance (Fig. 4B).
SCS livers had severe hepatic vacuolar degeneration, edema, and hepatic sinusoid congestion; BP livers had almost no vacuolar degeneration, hepatic sinusoid congestion, or inflammatory cell infiltration, and had less hepatic sinusoid congestion and hepatocyte edema than the P livers. The BP group had a significantly lower Suzuki score than the P and SCS groups (P < 0.05). In terms of DCD liver pathology, BMMSCs plus NMP was the best of the three storage methods, as it could improve liver pathology, and BMMSCs promoted NMP protection of the DCD liver (Fig. 4C).
NMP combined with BMMSCs attenuated DCD hepatocyte apoptosis
The Normal group had the fewest apoptotic cells, while the SCS group had the most. The P and PB groups had significantly fewer apoptotic cells than the SCS group; the PB group had fewer apoptotic cells than the P group (P < 0.05). This suggested that NMP and combined with BMMSCs could attenuate DCD hepatocyte apoptosis, and NMP combined with BMMSCs was superior to NMP alone and SCS (Fig. 4C).
NMP combined with BMMSCs improved DCD liver oxidative stress injury
MPO is a peroxidase, and is the main enzyme responsible for producing oxygen-free radicals. MDA is the final product of free radicals acting on lipids after peroxidation, and will affect the mitochondrial respiratory chain complex and key enzyme activities in the mitochondria. GSH is an important antioxidant and free radical scavenger in the body. Thus, MPO, MDA, and GSH are biomarkers of oxidative stress. IF and western blotting were used to detect the levels of MPO in liver tissue. The level of MPO in the PB group was significantly lower than that in the P and SCS groups, and the level of MPO in the P group was significantly lower than that in SCS group (P < 0.05) (Fig. 5A, B). The MDA levels in PB and P groups were significantly lower than that in the SCS group (P < 0.05) (Fig. 5C). GSH levels in PB and P groups were significantly higher than that in the SCS groups (P < 0.05) (Fig. 5D). These results suggested that NMP alone and combined with BMMSCs could reduce DCD liver oxidative stress injury, and NMP combined with BMMSCs was better than NMP alone.
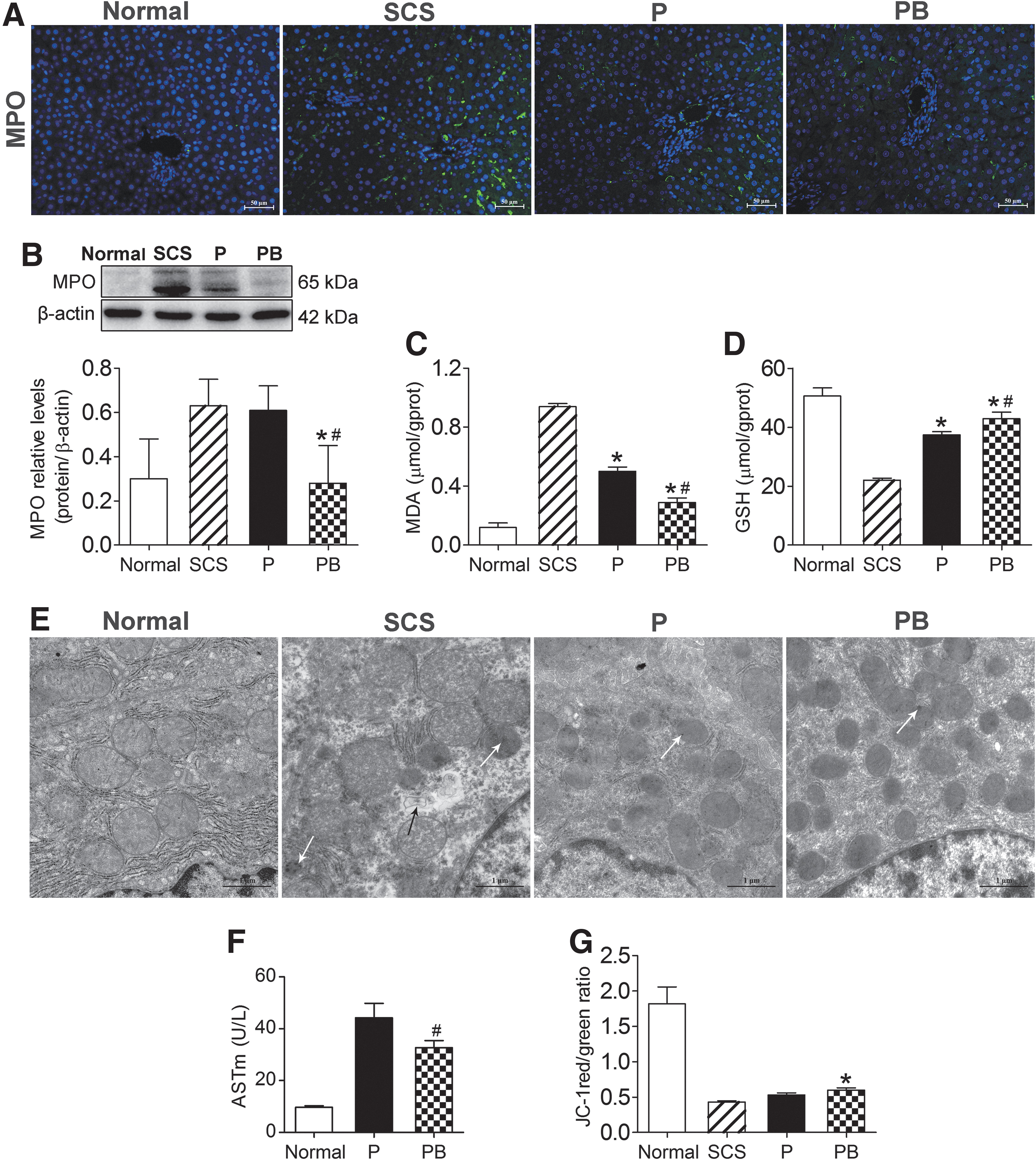
BMMSCs improved DCD liver oxidative stress, and repaired mitochondrial damage and function.
NMP combined with BMMSCs repaired DCD liver mitochondrial damage and improved its function
Transmission electron microscopy showed severe mitochondrial edema, evident vacuolization, irreversible mitochondrial damage, partial mitochondria dissolution, and disrupted mitochondrial cristae (most disappeared) in the SCS group. The BP group and P group showed almost no swelling, no vacuolization, the mitochondrial crest structure was intact, and there was less irreversible mitochondrial damage (Fig. 5E). As a mitochondrial damage marker, the ASTm level was significantly lower in the PB group than in the P group (P < 0.05) (Fig. 5F), suggesting that NMP combined with BMMSCs could ameliorate mitochondrial damage.
The mitochondrial membrane potential in DCD livers was significantly lower than that of normal livers, while the mitochondrial membrane potential level of the PB group was significantly higher than that of the SCS group (P < 0.05) and the mitochondrial membrane potential of the P group was lower than that of the PB group (Fig. 5G), which suggested that NMP combined with BMMSCs significantly improved the mitochondrial membrane potential of the DCD liver, which was superior to SCS and NMP alone.
Effects of BMMSCs on IAR20 cells after oxidative stress in vitro
Establishment of IAR20 oxidative stress injury model in vitro
To mimic the oxidative stress injury of the DCD liver during ischemia and reperfusion, we established an IAR20 cell oxidative stress model and observed the effects of BMMSCs on IAR20 cells after oxidative stress. With increasing H2O2 concentrations, cell stress injury gradually increased, and cell deformation, shedding, and necrosis appeared (Fig. 6A). ALT and AST levels gradually increased. The MDA level of IAR20 cells increased, and the level of GSH decreased, suggesting that the degree of oxidative stress injury gradually increased with increasing H2O2 concentration. The CCK-8 assay results showed decreased cell viability (Fig. 6B).
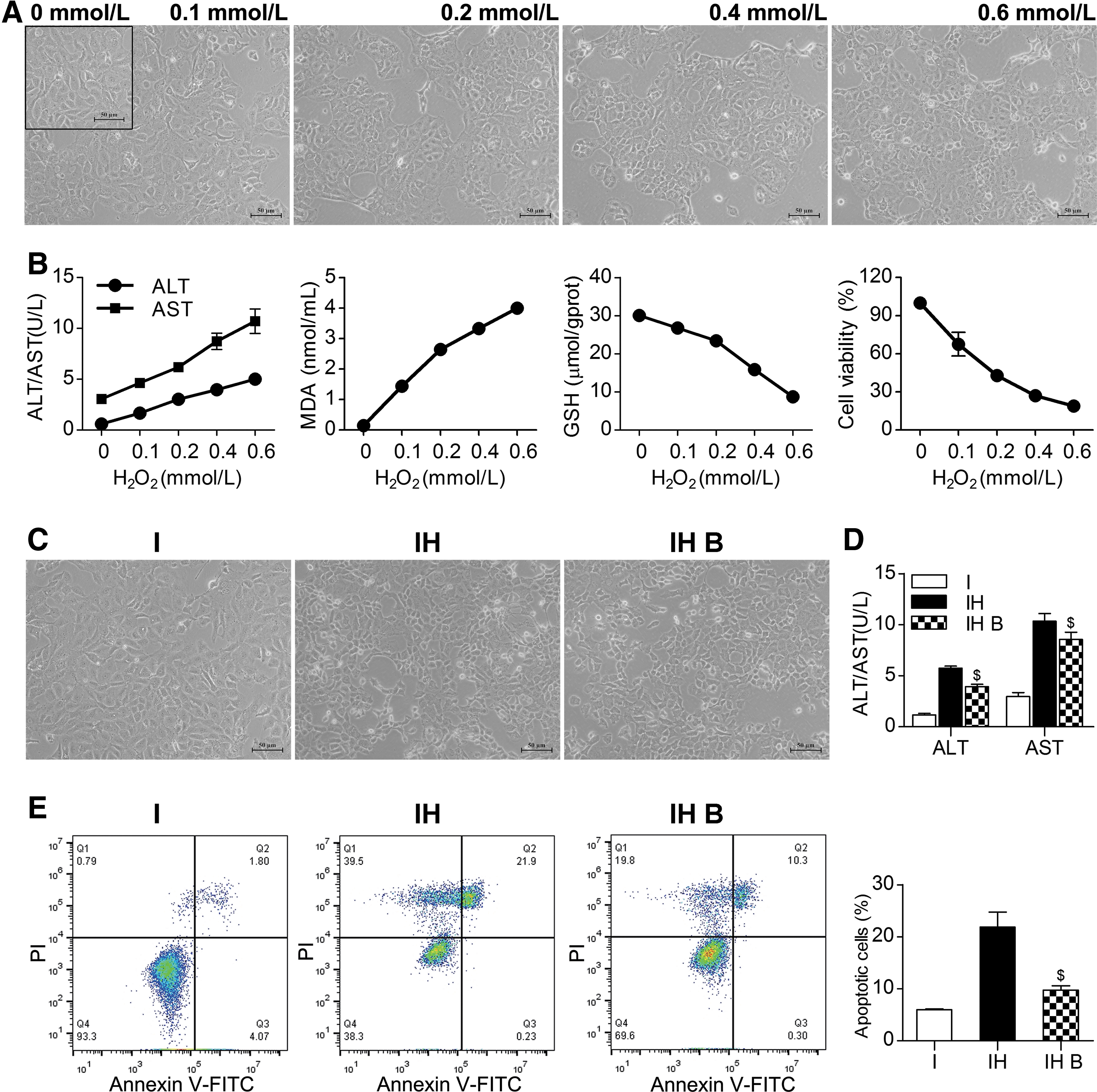
BMMSCs reduced damage and apoptosis of IAR20 cells after oxidative stress.
BMMSCs alleviated injury and apoptosis of IAR20 cells after oxidative stress
Cells in the IH B group had a better morphology, and there were significantly fewer necrotic cells than those in the IH group (Fig. 6C). The levels of ALT and AST in group IH B were significantly lower than those in IH group (P < 0.05) (Fig. 6D). The results of flow cytometry detection showed that the number of apoptotic cells in the IH B group was significantly lower than that in the IH group (P < 0.05) (Fig. 6E). This suggested that BMMSCs could reduce the injury and apoptosis of IAR20 cells after oxidative stress.
Effects of BMMSCs on reducing oxidative stress injury
IF and western blotting showed that the MPO level in the IH B group was significantly lower than that in the IH group (P < 0.05) (Fig. 7A, B). The MDA level in the IH B group was significantly lower than that in the IH group (P < 0.05) (Fig. 7C). The GSH level in the IH B group was significantly higher than that in the IH group (P < 0.05) (Fig. 7D). The release of ROS in the IH group increased significantly. The ROS level in the IH B group was significantly lower than that in the IH group, showing that ROS release was significantly suppressed (P < 0.05) (Fig. 7E). These results indicated a lower degree of IAR20 cell oxidative stress injury in the BMMSC coculture group, and that BMMSCs could ameliorate IAR20 cell oxidative stress injury.
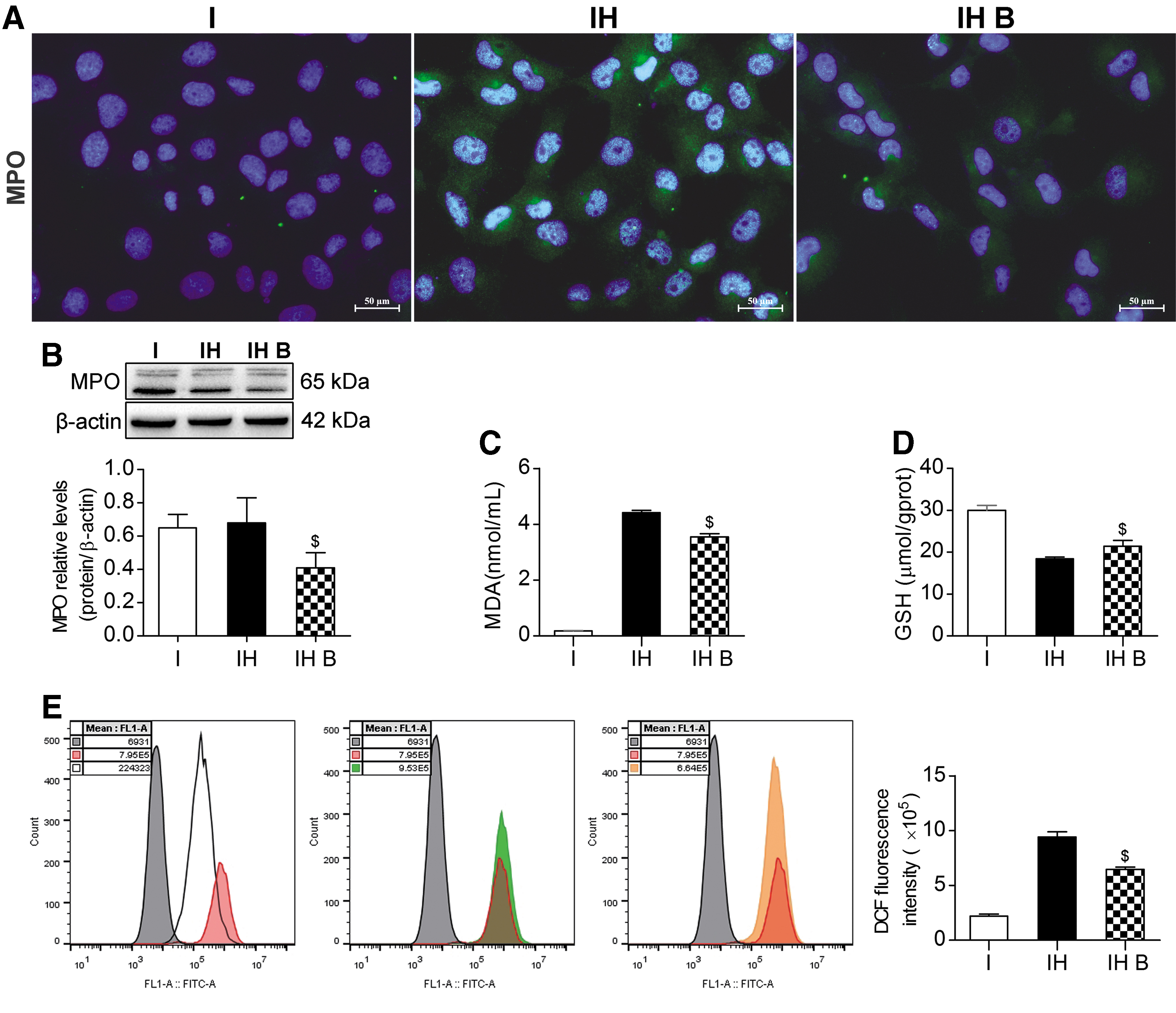
BMMSCs reduced oxidative stress injury.
Effects of BMMSCs on mitochondrial damage and function in IAR20 cells after oxidative stress
Transmission electron microscopy showed severe mitochondrial edema in the IH group. Most of the mitochondria were dissolved and necrotic and mitochondrial cristae had almost no normal structure. In the IH B group, mitochondrial edema was lighter, and there was less mitochondrial cristae structure disorder and necrosis in the IH B group than in the IH group (Fig. 8A). Mitochondrial injury marker ASTm levels were significantly lower in the IH B group than in the IH group (P < 0.05) (Fig. 8B), which suggested that BMMSCs could ameliorate mitochondrial damage after oxidative stress.
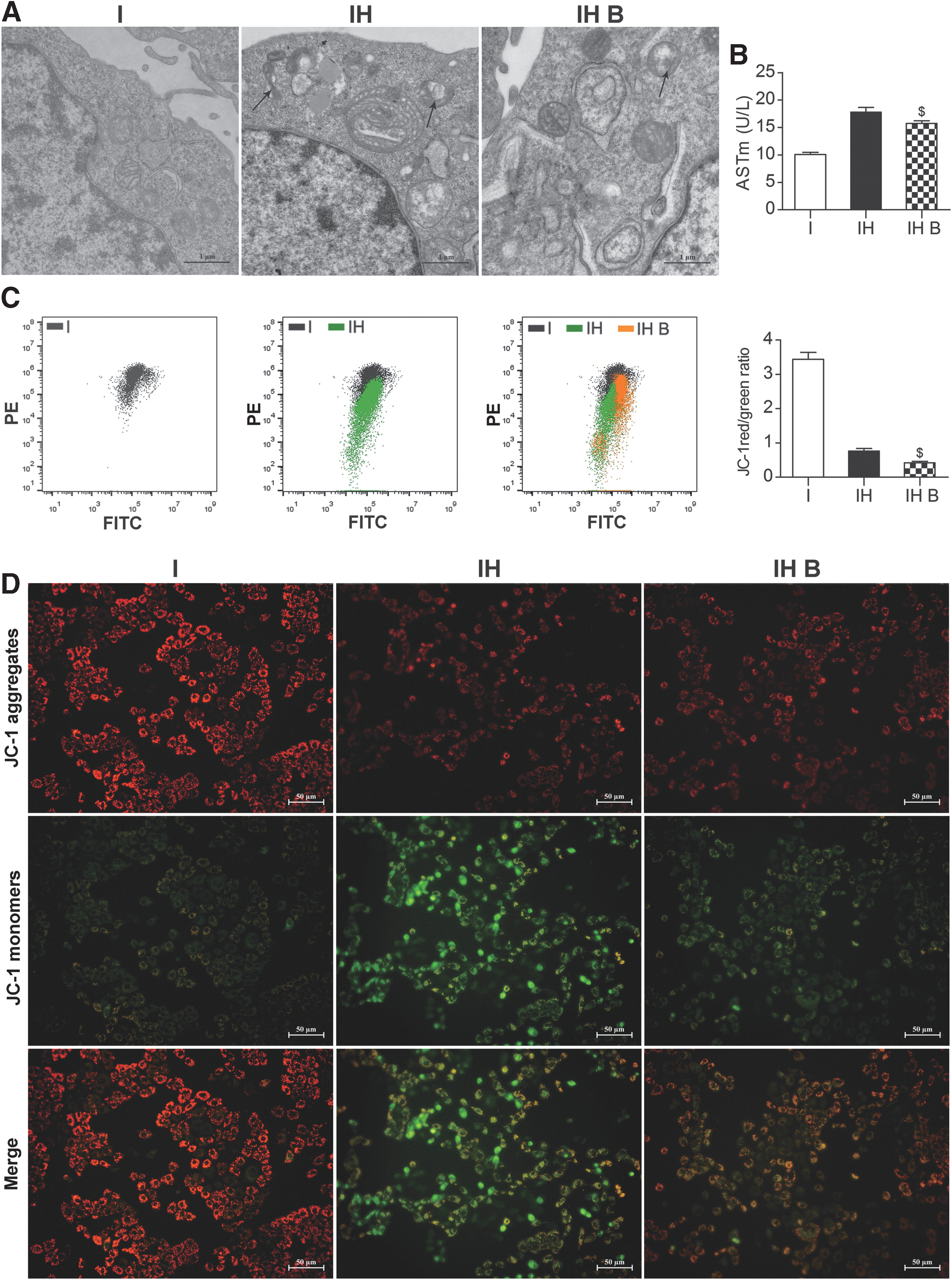
BMMSCs improved mitochondrial damage and function in IAR20 cell after oxidative stress.
Mitochondrial membrane potential detection showed that the proportion of mitochondrial JC-1 aggregates and monomers in the IH B group was significantly higher than that in the IH group, suggesting that the mitochondrial membrane potential was higher in the IH B group (P < 0.05) (Fig. 8C). Fluorescence microscopy showed that the fluorescence intensity of the mitochondrial JC-1 aggregates in the IH B group was significantly higher than that in the IH group (Fig. 8D). These results suggested that BMMSCs could improve the mitochondrial membrane potential function after oxidative stress injury.
Mechanism of BMMSCs' effects on IAR20 cell injury after oxidative stress
JNK, a subfamily of MAPK and part of the MAPK cascade, can be induced by various stresses or cytokines. JNK responds to various stress stimulations and induces NF-κB activation. NF-κB is a downstream signaling molecule of JNK. Detection of the proteins of the JNK-NF-κB signaling pathway in IAR20 cells after oxidative stress showed that the phosphorylation of JNK and NF-κB proteins in the IH B group was reduced significantly (P < 0.05), and JNK-NF-κB signaling pathway activation in group IH B was significantly inhibited, suggesting that BMMSCs inhibited the JNK-NF-κB signaling pathway after oxidative stress (Fig. 9A).
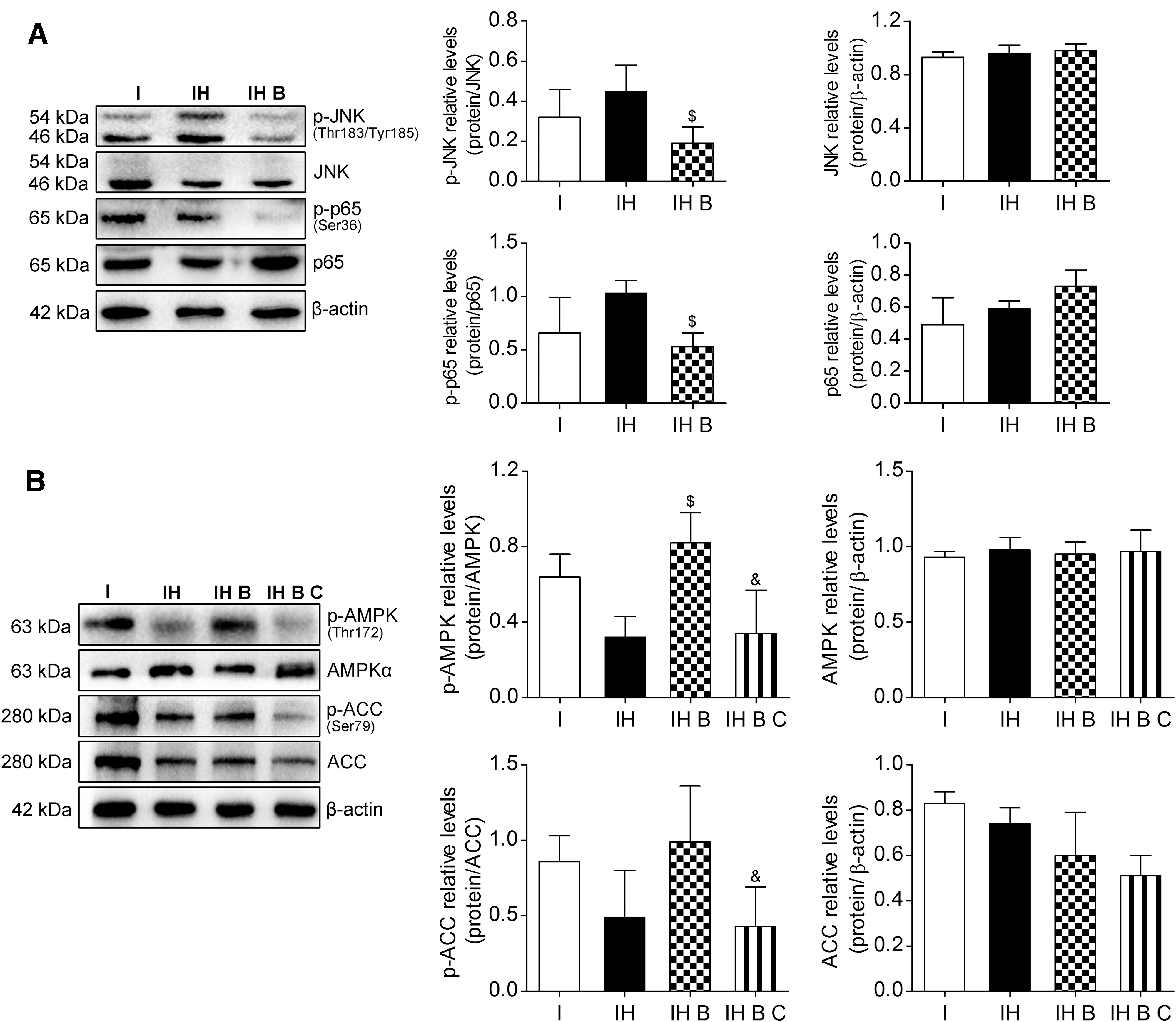
BMMSCs inhibited the JNK-NF-κB pathway and promoted AMPK activation in IAR20 cell after oxidative stress.
AMPK activation is regulated by metabolic stresses, such as glucose deficiency, hypoxia, ischemia, and metabolic toxins, and is a downstream target protein of ROS. The AMPK phosphorylation levels in the IH B and IH groups increased significantly (P < 0.05). The marker of the downstream AMPK phosphorylation level, p-ACC, also increased. After adding the AMPK inhibitor Compound C (15 μM), there was no significant difference in the total protein level of AMPK in the IH B C group; however, the level of p-AMPK decreased significantly (P < 0.05) (Fig. 9B).
Validation of the protective mechanism of NMP combined with BMMSCs on DCD liver
Finally, we verified the changes in JNK-NF-κB and AMPK in the DCD liver, and found that the phosphorylation of JNK and NF-κB proteins in the PB group decreased significantly (P < 0.05) (Fig. 10A). The activation of the JNK-NF-κB signaling pathway was significantly inhibited. We also found that the activation of liver AMPK was inhibited in the SCS group; however, PB significantly promoted the phosphorylation of AMPK and its downstream protein, ACC (P < 0.05) (Fig. 10B). These results suggested that during oxidative stress injury, BMMSCs inhibited the activation of the JNK-NF-κB signaling pathway and promoted AMPK activation in the DCD liver, which was consistent with the changes observed in the cell model.
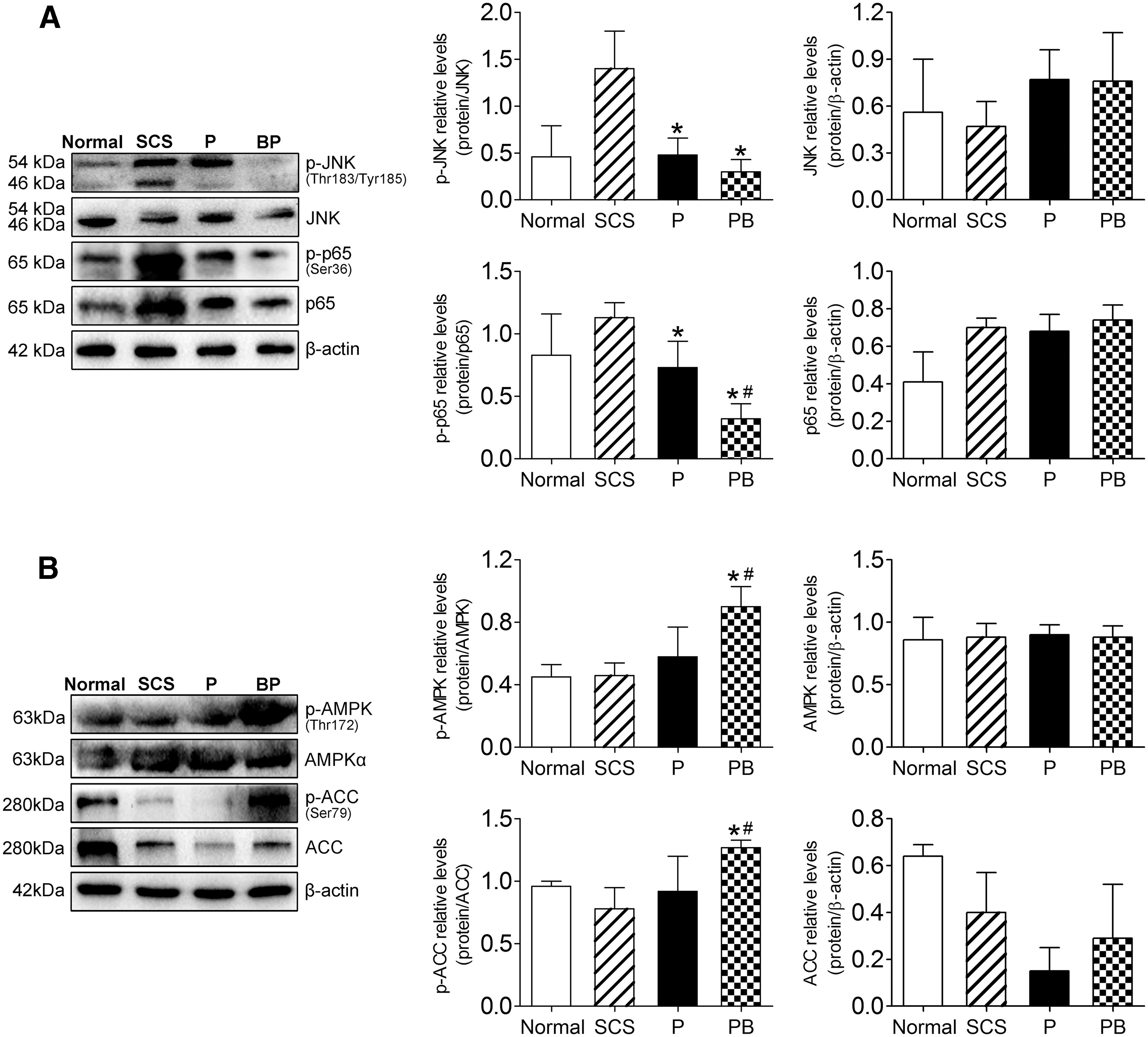
BMMSCs inhibited JNK-NF-κB activation and promoted AMPK activation in DCD liver.
Discussion
The DCD donor liver does not have sufficient physiological reserves to tolerate SCS-related injuries; therefore, liver mitochondrial function and energy metabolism are easily damaged, and are particularly sensitive to SCS-related IRI, which limits the use of SCS for the preservation of DCD donor livers [5,6]. The emergence of NMP has greatly improved the quality of DCD livers. Studies have shown that the NMP preservation method is superior to SCS [26], effectively preventing the liver damage caused by cold ischemia, promoting the elimination of liver metabolic waste and inflammatory factors, and improving liver energy metabolism. NMP allows the assessment of organ function before transplantation and significantly improves DCD liver IRI, donor liver activity, and the survival rate, thus effectively increasing liver utilization [27]. However, there are still many uncertainties, and this perfusion system requires further research and optimization to improve the quality of DCD livers [9,28,29].
In the present study, we used a stable single-cycle NMP system, in which the perfusion times were at the instant of perfusion and 2, 4, 6, and 8 h. Liver function, lactate clearance, and bile production were detected. Hepatic IRI improved gradually after perfusion, and the pathological manifestation was best at 4 and 6 h. Consistent with the liver function results, hepatocyte edema, hepatic sinusoid congestion, and other pathological manifestations deteriorated after 6 h. We evaluated the effect of the NMP system on the DCD liver quality comprehensively, and observed that livers from hour 6 had the best quality following NMP preservation. BMMSCs can regulate immunity, participate in anti-inflammatory activities, and secrete cytokines and exosomes, which contribute to their induced reduction in liver cell damage, acceleration of liver regeneration [22,30 –33], significant improvement of IRI [34], and reduced oxidative stress injury [35,36]. We confirmed that when NMP was combined with BMMSCs, the BMMSCs colonized the DCD liver and exerted the above-mentioned effects on the DCD liver. Therefore, we used NMP combined with BMMSCs to explore the impact on the quality of the DCD liver and the mechanism of the observed effects.
We evaluated the effects of NMP alone and NMP combined with BMMSCs on the quality of DCD livers. We found that with a perfusion time of up to 6 h, NMP combined with BMMSCs was significantly better than NMP alone in terms of improving liver function and promoting lactate clearance and bile secretion. In addition, NMP combined with BMMSCs significantly improved the liver pathology and IRI, and reduced hepatocyte apoptosis. These results verified that BMMSCs could increase the protective effect of NMP on DCD donor livers [22,37].
Hepatic IRI is a complex pathophysiological process involving oxidative stress [38,39], complement activation [40], and apoptosis [41]. In the early stage of IRI, Kupffer cells are activated, resulting in a large amount of ROS, proinflammatory factors, and cytokines [42 –45]. Inflammatory mediators increase the oxidative stress injury of cells, which is a characteristic of excessive ROS production [46 –48]. IRI occurs when the blood supply to the organ is interrupted and then restored. Although reperfusion of ischemic tissue is critical for survival, it can also cause mitochondrial dysfunction, oxidative damage, and cell death through excessive production of mitochondrial ROS, such that ROS are the key medium of IRI [49,50]. In vitro models showed that increased ROS levels could be observed during both warm and cold ischemia; however, the increase in ROS during warm ischemia was more sustainable than that during cold ischemia. In addition, extracellular ROS produced by Kupffer cells also damage mediators of IRI [51 –53]. A standard donor liver become expanded because of lower antioxidant defense capacity or higher ROS produced by the mitochondrial xanthine and xanthine oxidase system, which is more sensitive to lipid peroxidation [45,54]. A comprehensive consideration of these adverse factors in the DCD liver indicated that it is necessary to improve the overoxidative stress response of the DCD liver and its mitochondrial function [4]. Therefore, under different preservation methods, we tested the MPO, MDA, and GSH indexes related to oxidative stress injury of the DCD liver [55], and investigated the impact of NMP combined with BMMSCs on the DCD liver. We found that NMP combined with BMMSCs significantly reduced the levels of MPO and MDA, inhibited the release of ROS, and increased the production of GSH compared with the degree of liver stress injury in the SCS and NMP alone groups. Thus, NMP combined with BMMSCs plays an important role in ameliorating DCD liver oxidative stress injury. MSCs can inhibit the macrophage-mediated inflammatory response [32] and improve IRI [34]. Inhibition of MSCs on KCs activation and reduction of ROS production may also be part of their reparative effect.
During liver IRI, especially during warm ischemia, hepatocyte mitochondria are the earliest to show changes; mitochondria are very sensitive to hypoxia and oxidative stress, and are susceptible to ROS damage [56,57]. In early stage of liver reperfusion, irreversible mitochondrial oxidative damage first appears, Kupffer and endothelial cells have a higher degree of activation and a relatively higher production of ROS in NMP compared with that in hypothermic perfusion, which is prone to causing hepatocyte mitochondrial damage and is mainly related to higher proton dynamics at higher temperatures [12,58]. We found that the degree of mitochondrial damage in DCD livers under NMP combined with BMMSCs was lower, and the mitochondrial membrane potential improved significantly compared with that in the SCS and NMP alone groups. NMP combined with BMMSCs plays a significant protective role in reducing DCD liver mitochondrial damage and improving mitochondrial function, which could counteract the damage caused to mitochondria during early normothermic perfusion.
During warm ischemia, hypoxia leads to further oxidative stress and decreased mitochondrial biological function in the DCD liver [59]. A sudden increase in O2 content after reperfusion will produce mitochondrial ROS bursts and cause oxidative stress injury [60]. To further verify the protective mechanism of NMP combined with BMMSCs, we simulated the oxidative stress injury of the DCD liver during ischemia and reperfusion, and constructed a cell-level oxidative stress injury model in vitro to observe the mechanism of BMMSCs on oxidative stress. According to most previous studies of oxidative damage cell models, the degree of damage is usually controlled at about 50% of cell activity [61,62]. Accordingly, we choose a concentration of 0.2 mmol/L H2O2 for the construction of the IAR20 cell oxidative stress model in vitro. Cell model verification showed that the addition of BMMSCs significantly reduced the damage and apoptosis of IAR20 cells after oxidative stress stimulation, ameliorated mitochondrial damage, and maintained the mitochondrial membrane potential, which was consistent with the protective effect of BMMSCs on the DCD liver under NMP.
Studies have shown that JNK activation is closely related to ROS production. Prolonged JNK activation is ROS-dependent, and is associated with high levels of ROS inactivating phosphatases [63]. JNK plays a central role in ROS accumulation, in which ROS promotes JNK activation, and activation of the JNK pathway induces the accumulation of ROS; thus, JNK is activated via positive feedback [64]. We detected JNK in IAR20 cells after oxidative stress and observed significantly downregulated JNK activation under BMMSC coculture, and reduced activation of the downstream molecule, NF-κB [65,66]. Thus, BMMSCs inhibit the activation of JNK-NF-κB signaling pathway after oxidative stress.
JNK is a subfamily of MAPK and is part of the MAPK cascade. It is induced by various stresses or cytokines and is closely related to ROS. JNK induces NF-κB activation in response to various stress stimuli and NF-κB is a downstream signal of JNK [63,67]. AMPK, another signaling protein that is closely related to ROS, is an energy state sensor that can maintain the energy homeostasis of cells. Activation of AMPK is regulated by metabolic stresses, such as glucose deficiency, hypoxia, ischemia, and metabolic toxins, which in turn interferes with ATP metabolism [68,69]. AMPK is also a downstream target protein of ROS [70,71]. Once the liver returns to perfusion after ischemia, a sudden increase in the O2 content leads to a mitochondrial ROS outbreak, coupled with an increase in Ca2+ and a low pH value, which leads to the opening of mitochondrial permeability transition pores and causes mitochondrial damage and apoptosis [60]. Studies have shown that activation of the AMPK pathway in cells is significantly inhibited during oxidative stress injury, which indicates mitochondrial oxidative stress injury [72,73]. AMPK activation inhibits oxidative stress-mediated mitochondrial ROS production and ameliorates oxidative stress injury [74]. We detected that under BMMSC coculture, the phosphorylation level of AMPK was significantly upregulated in the IAR20 cells stimulated by oxidative stress, and the phosphorylation level of the downstream molecule ACC [70] was also upregulated; however, the difference was not significant, and other molecules may also inhibit the activation of ACC. To further verify the effect of BMMSCs on AMPK activation, we added AMPK inhibitors to the BMMSCs coculture group to block AMPK activity, which resulted in significantly downregulated AMPK phosphorylation. Finally, comparing the phosphorylation levels of AMPK in the three groups indicated that BMMSCs promoted AMPK activation after oxidative stress. In addition, while BMMSCs promote the activation of AMPK, AMPK could also inhibit the activation of the JNK-NF-κB pathway, reduce the production of ROS, and reduce oxidative stress injury [74]. In this respect, our results are consistent with those of previous reports.
Finally, we detected the levels of JNK-NF-κB and AMPK in DCD livers under different storage conditions and found that the activation level of the JNK-NF-κB pathway in liver was significantly downregulated under NMP alone and combined BMMSCs; the activation of AMPK in NMP combined BMMSCs was significantly upregulated compared with that in the SCS and NMP alone groups. AMPK activation in the NMP liver was upregulated compared with that in the SCS liver, but the difference was not significant. The mechanism of the effect of BMMSCs in NMP on the DCD liver was basically consistent with the results of the cell model. In short, NMP combined with BMMSCs ameliorates oxidative stress injury of the DCD liver by inhibiting the activation of the JNK-NF-κB signaling pathway and promotes AMPK activation to reduce mitochondrial damage. This further validates the protective effect of this system on oxidative stress response and the mitochondrial function of the DCD liver.
It should be noted that the present study still has some shortcomings; for example, the NMP system is a single circulation system without a drainage device. In addition, excessive perfusion time limits the level of organ repair. In future research, we will improve the perfusion method. Finally, in the present study, we did not verify whether the donor liver under these conditions could achieve the desired in vivo results, which will also be our future research direction.
Conclusion
In the context of a shortage in liver donors, the DCD liver is an effective method to expand the available donor pool. To solve the problem of poor DCD liver quality, NMP combined with BMMSCs could reduce oxidative stress by inhibiting the JNK-NF-κB pathway, promoting AMPK activation, protecting damaged mitochondria, and improving oxidative stress injury and mitochondrial function in rat DCD livers. The present study identified protective factors and provided experimental evidence for the clinical use of DCD livers. We analyzed the improvement of DCD liver quality induced by NMP combined with BMMSCs pretransplantation; however, assessing recipients' quality of life after transplantation would be more important. Therefore, our future research direction is to assess whether the NMP-based preservation system can improve a transplant recipient's quality of life or prolong their survival.
Footnotes
Acknowledgments
The authors thank the teachers at the NHC Key Laboratory of Critical Care Medicine and the Tianjin Key Laboratory of Organ Transplantation for their technical support.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The work was supported by National Natural Science Foundation of China (grant nos. 81670574, 81441022, and 81270528) and the Natural Science Foundation of Tianjin, China (grant nos. 08JCYBJC08400, 11JCZDJC27800 and 12JCZDJC25200).