
Abstract
Head and neck squamous cell carcinomas (HNSCCs) are malignancies that originate in the mucosal lining of the upper aerodigestive tract. Despite advances in therapeutic interventions, survival rates among HNSCC patients have remained static for years. Cancer stem cells (CSCs) are tumor-initiating cells that are highly resistant to treatment, and are hypothesized to contribute to a significant fraction of tumor recurrences. Consequently, further investigations of how CSCs mediate recurrence may provide insights into novel druggable targets. A key element of recurrence involves the tumor's ability to evade immunosurveillance. Recent published reports suggest that CSCs possess immunosuppressive properties, however, the underlying mechanism have yet to be fully elucidated. To date, most groups have focused on the role of CSC-derived secretory proteins, such as cytokines and growth factors. Here, we review the established immunoregulatory role of exosomes derived from mixed tumor cell populations, and propose further study of CSC-derived exosomes may be warranted. Such studies may yield novel insights into new druggable targets, or lay the foundation for future exosome-based diagnostics.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common type of cancer, with ∼500,000 new cases each year worldwide [1,2]. Over 70% of head and neck cancers are associated with alcohol and tobacco consumption [3,4]. HNSCC early-stage treatment typically includes surgery or radiotherapy while advance-stages frequently employ a combination of surgery followed by chemoradiotherapy [5,6]. Patient survival rates are often low as a result of local and regional recurrence, with a 40% mortality within 5 years post-treatment in high-risk patients [7 –10].
Immunosurveillance and immunoediting play an integral role in the progression and recurrence of HNSCC tumors [11,12]. The development of murine tumor models with molecularly defined immunodeficiencies has enabled the field to demonstrate the existence of cancer immunosurveillance processes that are capable of preventing tumor progression [13,14]. A large body of work has established that the immune system can select for the emergence of tumors with limited immunogenicity, capable of escaping immune recognition and elimination [15 –17]. This work led to the development of the cancer immunoediting hypothesis that determines the potential host-protective and tumor-sculpting properties of the immune system during tumor development [18,19].
The immune system recognizes tumors through the expression of tumor-associated antigens (TAA) [20,21]. TAAs are phagocytozed by professional antigen-presenting cells (APCs), such as macrophages and dendritic cells (DCs) [22,23]. APCs then present TAA on major histocompatibility complex (MHC)-II complexes, which subsequently interact with T cell receptors, thereby inducing an activation signaling cascade that results in the cytotoxic elimination of TAA bearing tumor cells [15,24].
Progressive tumors may evade and impair the immune system through the release of a variety of factors, creating a hypoimmunogenic tumor microenvironment [25,26]. The secretion of cytokines with anti-inflammatory properties such as transforming growth factor beta (TGFβ) and interleukin (IL)-10 can lead to the suppression of immune cells involved in antitumor immunity, such as CD8+ T cytotoxic cells and DCs [27 –35]. These cytokines can also induce immune cells with immunosuppressive properties such as regulatory T lymphocytes (Tregs) and myeloid derived suppressor cells (MDSCs), which permit tumors to evade immunosurveillance and elimination [36 –43].
In recent years, several groups have leveraged this increased understanding of the immunobiology underlying tumor development and immunoediting to develop novel targeted immunotherapeutic strategies to reactivate the immune system [44 –46]. The development of antagonist antibodies against immune checkpoint pathways have led to several successful drug approvals and effective implementation into clinical practice [47 –49].
Antibody inhibitors of the PD-1/PD-L1 axis is one such therapeutic approach that has achieved remarkable success in a subset of patients [50,51]. In addition, several novel immunotherapies developed from genetically modified chimeric antigen receptor T cells (CAR T cells), have shown significant promise in targeting hematological malignancies, such as certain subtypes of leukemia and lymphoma [52,53]. However, the application of CAR T cell technology to solid tumors remains challenging [54 –57]. Epidermal growth factor receptor (EGFR) has been identified as a potential target in epithelial tumors, such as HNSCC [58,59]. This has led to the development of EGFR targeting CAR T cells in non-small cell lung cancer that have undergone early stage clinical trials and has been determined to be safe [60,61]. Further elucidation of the mechanisms by which tumors develop equilibrium and eventually escape immunosurveillance is critical to develop additional novel therapeutic approaches and diagnostics [62,63].
Tumor-derived exosomes
To date, most reports investigating immunoediting by tumors has focused on canonical secretory factors, such as growth factors and cytokines [64,65]. However, there is a growing body of evidence that suggests that intercellular communication between tumor cells and stroma and immune cells also occurs through tumor-derived extracellular vesicles called exosomes (TEX) [66 –69]. Exosomes are cellularly secreted lipid bilayer nanovesicles secreted by virtually all cells [70,71]. Exosomes function as a robust and intricate intercellular communication system, which is evolutionarily conserved down to gram negative bacteria [72,73]. TEX transmit malignant information in the form of proteins, RNAs, lipids, and metabolites that can reprogram recipient cells [74,75].
There has been a growing body of work indicating that TEX isolated from the plasma of cancer patients may hold value as diagnostic and prognostic biomarkers, as a “liquid biopsy” of the tumor [76 –78]. Exosomes' contents and concentration in the plasma of patients with HNSCC has allowed for the differentiation between patients with active disease, no evident disease, and between early/advanced stages of tumor progression [79,80]. These findings highlight the potential use of TEX diagnostic biomarkers for HNSCC [81,82]. Indeed, such TEX-based biomarkers as liquid biopsy diagnostics from Exosome Diagnostics were recently approved for clinical use for prostate and lung cancer [83,84]. These diagnostics aid clinicians in detecting, diagnosing, and monitoring cancer progression in addition to identifying the unique genetic composition of each patient's tumor.
Several groups have also established that TEX functionally mediate cancer progression via facilitating tumor growth and promotion of the tumor microenvironment through localized suppression of immune surveillance through a variety of reported mechanisms [85 –87]. APCs, such as macrophages and DCs, phagocytose TEX, initiating signaling cascades that ultimately induce a regulatory phenotype among professional APCs [88,89]. Ligands on the surface of TEX can also bind cognate receptors on a cell's surface, inducing specific pathway signaling activation, which can also result in receptor-mediated endocytosis, releasing the contents of TEX within the target cell populations [90,91]. Alternatively, TEX ligands can bind cell surface receptors, like MHC-I on lymphocytes, triggering suppressive signaling pathways without entering the cell [92,93]. Further, PD-L1 expression on TEX has been demonstrated to suppress antitumor immunity and memory, and it may potentially account for limited response rates of current therapeutic antibody approaches [94].
TEX can also suppress the immune system indirectly through ectoenzymes CD39 and CD73 bound to the surface of TEX. CD39 hydrolyzes ATP into 5′ AMP and CD73 hydrolyzes 5′ AMP into adenosine [95,96]. Adenosine signals through the A2AR and A2BR receptors on tumor-associated macrophages (TAMs), MDSCs, natural killer cells, Tregs, DCs, and cytotoxic CD8+ T cells in a paracrine fashion (Fig. 1) [97 –100]. HNSCC-derived TEX have also been shown to induce apoptosis of CD8+ cytotoxic T cells [101,102]. CD95 and PD-1 receptors on the surface of CD8+ T cells are bound by TEX surface molecules FasL and PD-L1, respectively, inducing apoptosis [103,104]. Additionally, excess levels of PD-L1+ exosomes detected in patient plasma were associated with advanced HNSCC disease [104]. TAMs are highly abundant in the tumor microenvironment and have been established to promote tumor progression via facilitating immune escape [105,106]. Recently, miR-21+ exosomes from SNAIL+ cancer cells have been demonstrated to polarize macrophages to the M2 phenotype by targeting PDCD4 and IL-12A [107]. These studies demonstrate that TEX are capable of creating an immunosuppressive microenvironment via elimination of CD8+ cytotoxic T cells and differentiation of TAMs toward an M2 regulatory phenotype.
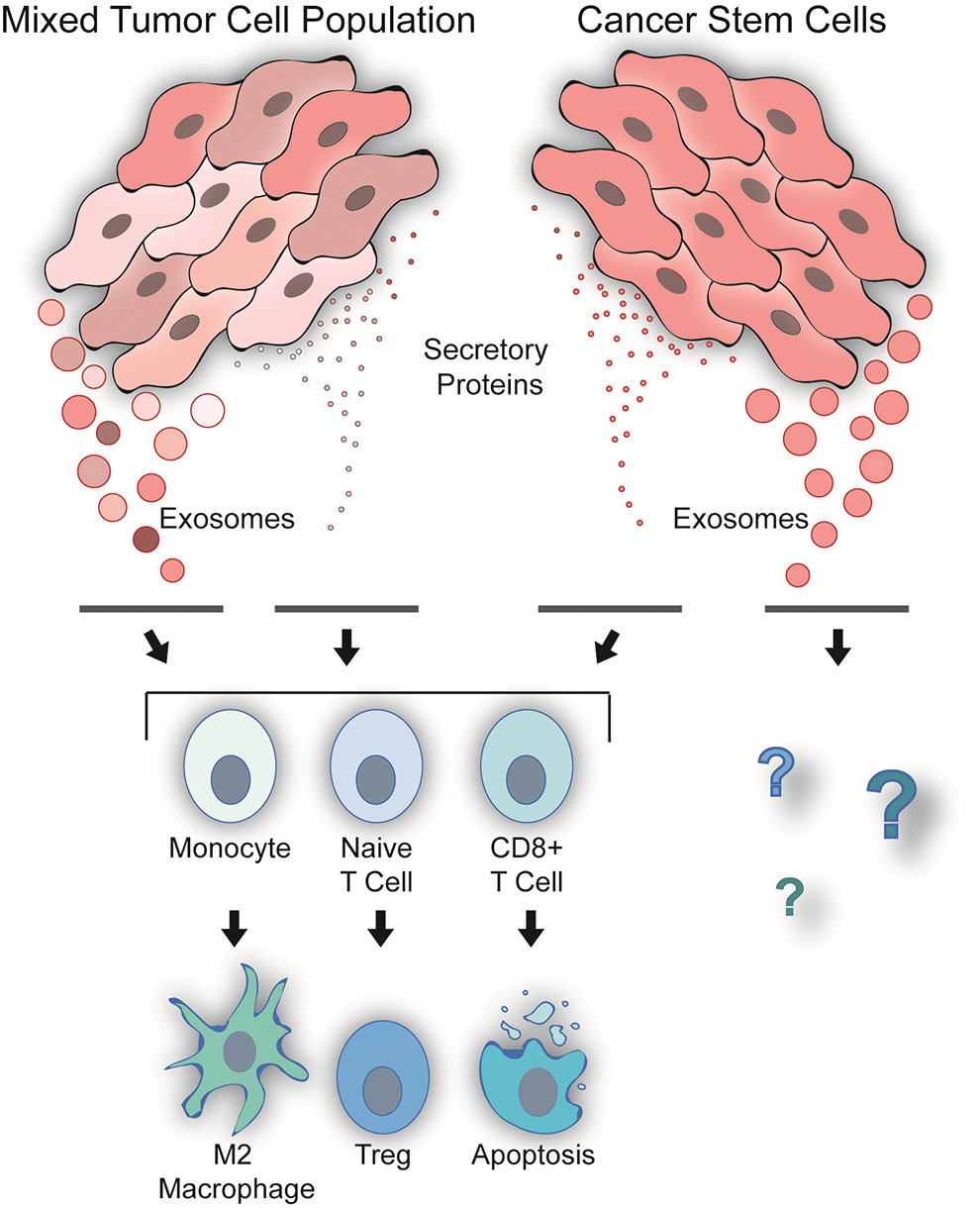
Exosomes derived from mixed tumor cell populations possess potent immunoregulatory properties, including, M2 macrophage polarization, Treg induction, and elimination of CD8+ cytotoxic T cells. CSCs have been shown to have immunomodulatory properties, yet it is currently not clear if CSC derived exosomes mediate these properties. CSCs, cancer stem cells. Treg, regulatory T lymphocyte. Color images are available online.
Tregs are vital for maintaining tolerance, thwarting autoimmunity, and restraining chronic inflammatory diseases [108 –113]. However, Tregs also limit beneficial responses through the suppression of antitumor immunity [114,115]. Tregs possess several mechanisms at their disposal to mediate their immunosuppressive effects, including suppression by cytolysis, suppression by metabolic disruption, suppression by inhibitory cytokines, and suppression by the modulation of DC maturation or function [116,117].
Elevated levels of Tregs have been observed in multiple tumor types [118]. Tregs and conventional CD4+ T cells are recruited to the tumor microenvironment via CCL20 of TEX [119]. TEX induce CD4+CD25- T cells into CD4+CD25+FOXP3+ Tregs in a TGFβ and IL-10-dependent manner, which results in phosphorylation and activation of the Smad2/3 and STAT3 signaling factors [120]. TEX also induce the upregulation of several immunosuppressive genes COX2, IL-10, CD39, CD73, PD-L1, and CD26 in Tregs [121]. The subsequent secretion of TGFβ and IL-10 by Tregs suppresses the proliferation of antitumor T cells via modulation of DC function and maturation [122]. Tregs also secrete cytolysis via secretion of granzyme A, granzyme B, and perforin targeting cells responsible for immunosurveillance [123,124].
Cancer stem cells
Solid tumors are heterogeneous mixtures of different cellular subpopulations, which possess multiple phenotypes, differentiation, and mitogenic potentials [125 –127]. A small subpopulation of tumor cells have been demonstrated to increase tumorigenicity, with stem cell-like abilities to self-renew and differentiate, which have become known as cancer stem cells (CSCs) [128,129]. CSCs have been attributed to resistance seen against cancer therapies, surviving both chemo and radiotherapy treatment and giving rise to local or distal recurrence in patients (Fig. 2) [130,131]. Current cancer therapeutic interventions such as surgery, radiotherapy, and chemotherapy eliminate most tumor cells, yet residual therapy-resistant CSCs may account for recurrence [131 –133].
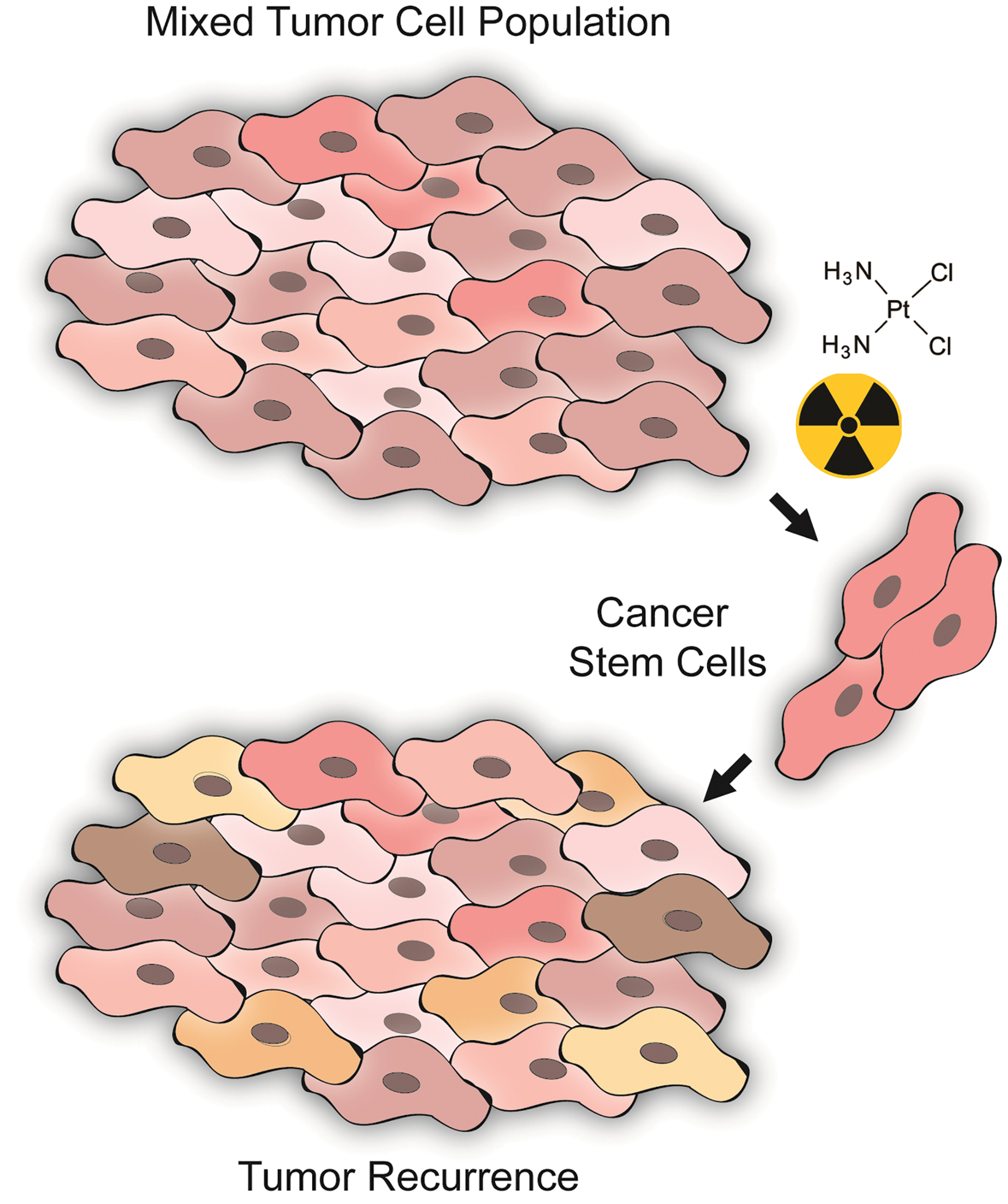
CSCs have been reported to possess chemoradiotherapy resistance properties that result in tumor recurrence. The determination of the mechanisms by which CSCs escape canonical therapeutic interventions, and subsequent evasion of immunosurveillance may provide insights into novel druggable targets designed to limit tumor recurrence. Color images are available online.
Al-Hajj et al. demonstrated that a population of tumor cells in breast cancer had the ability to self-renew and differentiate, and identified cell markers for CSCs, CD44+CD24- [128]. When CD44+CD24- CSCs were injected into mice, tumors formed that were phenotypically diverse and resembled the heterogeneous nature of the tumor the CSCs originated from [128]. The CD44- cells were not tumorigenic, cementing CD44 as an important marker for the identification of CSCs [129]. Similarly, CSCs were identified using these markers in HNSCC with similar results [134]. Populations of CD133+ cells have also been demonstrated to increase tumorigenesis, in a fashion similar to that of CD44+ cells [135]. Studies of CSC surface markers have shown CD44+ cell populations to more highly co-express CD133 than the CD44- cell population [133,136]. ALDH-1 was identified as an additional CSC marker, where CD44+CD24-ALDH-1+ cells displayed higher potential to form tumors and possessed increased radioresistance [137].
CSCs are essential for the recurrence, and several reports have established that they protect the developing tumor from immunosurveillance and elimination by promoting a protumor microenvironment and by regulating the immune system. Recently, Miao et al. established that adaptive immune resistance emerges from CSCs [126]. CSCs demonstrate stronger inhibition of CD8+ T cells and induction of Tregs and MDSCs than non-CSC tumor cell subpopulations [138]. Several reports have established that CSCs downregulate the presentation of MHC antigens, contributing to escape from CD8+ cytotoxic T lymphocytes [139]. CSCs have also been demonstrated to polarize TAMs toward the regulatory M2 phenotype [140].
To date, most published reports of the mechanisms underlying CSCs' immunosuppressive properties have focused on their secretion of various cytokines including TGFβ-1, and IL-6 [135,141,142]. These immunoregulatory cytokines possess potent anti-inflammatory properties, mediated by Tregs and MDSCs, which subsequently secrete additional cytokines with additional immunomodulatory properties that suppress the immune system, promoting a tumor permissive microenvironment.
CSCs are tumor-initiating cells whose progeny generate the bulk of subsequent tumor tissue. A growing body of work has established that exosomes derived from bulk tumor tissue are key mediators of communication within the tumor microenvironment, and induce protumorigenic effects in immune cells, and other nontumor bystander cells [143 –145].
To date, few published reports have investigated the physiology and functional properties of CSC secreted exosomes (CSCEX) [146 –148]. However, it is feasible that CSC secrete CSCEX, which possess similar immunosuppressive properties similar to TEX [85]. Numerous reports have established that exosomes derived from other stem cell source possess comparable properties as well [149 –152].
The determination of whether CSCEX allow for CSC to evade immunosurveillance and facilitate relapse within an immune-permissive tumor microenvironment represents a current critical gap in knowledge that warrants further investigation [153]. Better understanding of the molecular mechanisms by which CSCEX potentially regulate antitumor immunity may yield vital insights into tumor progression, and elucidate novel drug targets for aggressive tumors [66]. Such investigations may also yield insights into novel diagnostic biomarkers that are able to discern CSC-load post-treatment using CSCEX isolated from liquid biopsies. Taken together, there is strong evidence to suggest that further study of CSCEX may yield valuable biomedical and translational insights into recurrent tumor progression.
Conclusion
CSCs are highly tumorigenic and resistant to traditional cancer treatments, giving rise to local and distal tumor recurrence. To date, published reports have focused on CSCs' secretions of cytokines rather than exosomes. However, a growing body of evidence has established that exosomes derived from complex tumor tissue specimens comprised of multiple subpopulations of cells possess potent immunoregulatory properties that facilitate tumor progression. It is critical to distinguish whether these exosomes were derived from CSCs or solely from non-CSC to identify potential mechanistic differences for novel therapeutic interventions and diagnostics specifically targeting CSCs. However, it is feasible that many of the properties attributed to TEX are also associated with CSCEX. Further investigation of CSCEX, and their effects on the immune system, may uncover previously undiscovered mechanisms underlying the suppression of antitumor immunity, thereby elucidating novel druggable immunotherapy targets. Targeting of CSCs is an attractive approach to limit cancer treatment resistance, and to prevent tumor relapse.
Footnotes
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Authors are supported by the National Center for Advancing Translational Sciences, National Institutes of Health through grant number UL1 TR001860 and linked award KL2 TR001859. As well as, CIRM EDUC2–08390, the UC Office of the President's Multi-campus Research Program Grant (MRP-17-454909), STAIR Grant, STAIR-Plus Grant, CTSC Rapid Translational Grant (UL1-TR001860), T32 Cardio NIH T32-HL086350, Denny and Jeanene Dickenson Fellowship, NIH Transformative R01GM099688, NIH T32-GM008799.