
Abstract
As sites of cellular respiration and energy production, mitochondria play a central role in cell metabolism. Cell differentiation is associated with an increase in mitochondrial content and activity and with a metabolic shift toward increased oxidative phosphorylation activity. The opposite occurs during reprogramming of somatic cells into induced pluripotent stem cells. Studies have provided evidence of mitochondrial and metabolic changes during the differentiation of both embryonic and somatic (or adult) stem cells (SSCs), such as hematopoietic stem cells, mesenchymal stem cells, and tissue-specific progenitor cells. We thus propose to consider those mitochondrial and metabolic changes as hallmarks of differentiation processes. We review how mitochondrial biogenesis, dynamics, and function are directly involved in embryonic and SSC differentiation and how metabolic and sensing pathways connect mitochondria and metabolism with cell fate and pluripotency. Understanding the basis of the crosstalk between mitochondria and cell fate is of critical importance, given the promising application of stem cells in regenerative medicine. In addition to the development of novel strategies to improve the in vitro lineage-directed differentiation of stem cells, understanding the molecular basis of this interplay could lead to the identification of novel targets to improve the treatment of degenerative diseases.
Introduction
S
Given the promising applications of stem cells in regenerative medicine and cell therapy, there is increasing interest in understanding the mechanisms regulating their self-renewal, pluripotency, and plasticity. Recent data support strong and direct involvement of mitochondria and oxidative metabolism in the regulation of stem cell pluripotency [3]. Cells adapt the number and activity of mitochondria in response to environmental and cellular cues through biogenesis, turnover, and fusion and fission processes [4]. Besides playing a fundamental role in energy production through oxidative phosphorylation (OXPHOS), mitochondria play important roles in amino acid, fatty acid, and steroid metabolism, as well as in cell signaling by reactive oxygen species (ROS) production, calcium homeostasis, and apoptosis [4].
The current review highlights the mitochondrial and metabolic changes that are associated with the differentiation of stem cells and the underlying pathways. We first describe the mitochondrial remodeling that occurs during pluripotent stem cell (PSC) differentiation and reprogramming, as well as the evidence, obtained by interfering with mitochondrial function, suggesting that mitochondria participate actively in those processes. We overview the recent data emerging from SSC studies suggesting that mitochondrial biogenesis and metabolic switches may be hallmarks of cell differentiation processes. Next, we address 3 questions: (i) What advantages does glycolysis bestow on PSCs? (ii) Are the mitochondrial and metabolic remodelings early or late events of stem cell differentiation/reprogramming? (iii) Can mitochondrial dynamics regulate the fate of stem cells? Finally, we provide an overview of the elucidated and putative molecular actors underlying the crosstalk between mitochondria, metabolism, pluripotency, and differentiation.
Opposite Mitochondrial Remodeling and Metabolic Shifts During PSC Differentiation and Reprogramming
Mitochondria display a characteristic ultrastructure. However, the first observations of mouse and human ESCs (mESCs and hESCs) using transmission electron microscopy surprisingly revealed immature, rare, and globular mitochondria displaying a perinuclear localization [5 –10] and containing poorly developed cristae, as well as an electron-lucid matrix [11 –15]. In contrast, somatic cells such as fibroblasts show mature elongated mitochondria, with numerous cristae and an electron-dense matrix [9] (Fig. 1). During the in vitro differentiation of hESCs, elongation of the mitochondrial network and maturation of the cristae ultrastructure are observed [5,8,9,15]. The reprogramming of human and mouse somatic cells into iPSCs results in opposite remodeling of the mitochondrial network in a process known as mitochondrial rejuvenation [8,9,16]. The ultrastructure, morphology, and intracellular distribution of mitochondria undergo reversible changes during the reprogramming of fibroblasts into iPSCs as well as subsequent redifferentiation into fibroblast-like cells, which support the importance of mitochondrial remodeling in differentiation and reprogramming events [8].
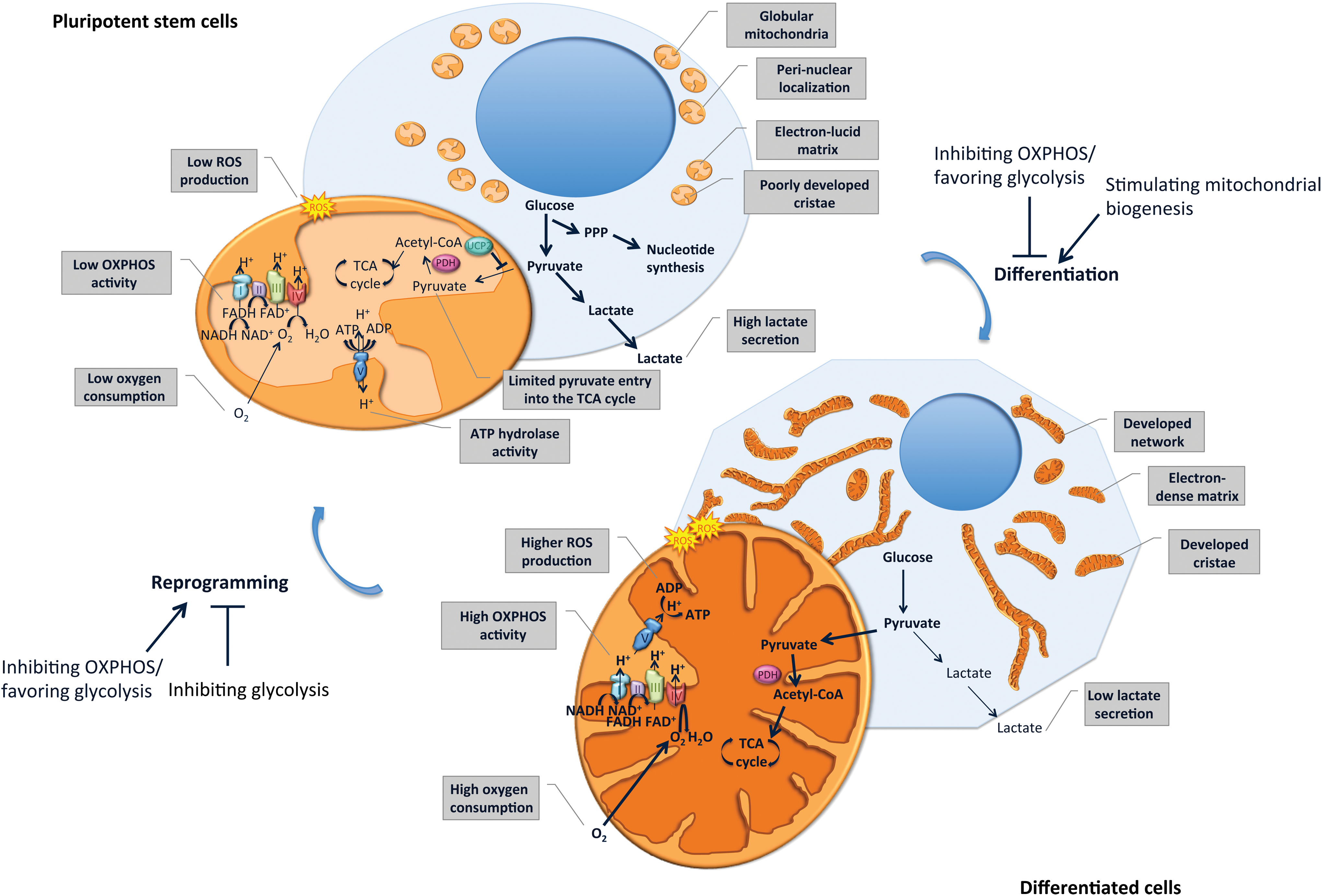
Mitochondria and metabolism remodeling upon pluripotent stem cell differentiation and reprogramming. Pluripotent stem cells display globular immature mitochondria, localized perinuclearly, characterized by an electron-lucid matrix and poorly developed cristae. Energy production in pluripotent stem cells is mainly generated by a high glycolysis rate, leading to increased lactate production, whereas oxidative phosphorylation (OXPHOS) activity is limited, leading to reduced oxygen consumption and reactive oxygen species (ROS) production. Although substrate entry into the tricarboxylic acid (TCA) cycle is limited, intermediates of glycolysis enter the pentose phosphate pathway (PPP) and serve as substrates for the nucleotide synthesis required to sustain self-renewal. In differentiated cells, a more developed mitochondrial network, characterized by a more electron-dense matrix and developed cristae, ensures ATP production through increased OXPHOS activity. This results in elevated oxygen consumption and ROS production and reduced production of lactate through glycolysis. Interestingly, inhibiting OXPHOS or stimulating glycolysis impedes stem cell differentiation while favoring reprogramming into induced pluripotent stem cells (iPSCs). In contrast, stimulating mitochondrial biogenesis favors cell differentiation, whereas inhibiting glycolysis impairs reprogramming. Color images available online at
Besides morphological and ultrastructural changes, studies have observed an increase in mitochondrial DNA (mtDNA) content [15,17] and mass [8] during the differentiation of hESCs, mESCs, and iPSCs. However, the evidence of a parallel increase in cell volume upon differentiation has moderated this apparent increase in mitochondrial content [18]. Further studies are needed to determine whether mitochondrial biogenesis reflects an adaptation to the increased cell volume upon differentiation.
In addition to the induction of mitochondrial biogenesis, the differentiation of PSCs, which collectively refer to ESCs and iPSCs, is associated with a metabolic shift, characterized by a transition from a predominant glycolysis-based metabolism in PSCs toward an increased OXPHOS-based metabolism in differentiated cells. Differentiated somatic cell lines display increased oxygen consumption rates, respiratory reserve capacity, and intracellular ATP content while secreting less lactate, indicative of reduced glycolysis, in comparison with hPSC lines [9]. Accordingly, the differentiation of hESCs, mESCs, and iPSCs is associated with increased intracellular ATP levels and lower lactate production, while reprogramming of somatic cells into iPSCs is accompanied by the reverse modifications [8,15,16,19]. Proteome remodeling, characterized by the induction of glycolytic enzymes and concomitant downregulation of multiple subunits of the mitochondrial complexes I and II, occurs during the reprogramming of mouse somatic cells into iPSCs [16].
Intracellular ATP levels of PSCs are more sensitive to inhibition of glycolysis than to inhibition of OXPHOS, whereas the opposite is observed in differentiated cells [18]. This observation supports glycolysis playing a predominant role in ATP production in hPSCs. Accordingly, the inhibition of OXPHOS with oligomycin, an F1F0 ATP synthase inhibitor, decreases intracellular ATP by less than 5% in hPSCs, suggesting that minimal amounts of ATP are produced by OXPHOS in PSCs [18]. However, inducing mitochondrial uncoupling in hESCs, mESCs, and iPSCs using CCCP (carbonyl cyanide m-chlorophenylhydrazone) results in a drop in intracellular ATP levels and reduces proliferation rates. The AMP/ATP ratio increases in mESCs treated with the complex III inhibitor, antimycin A, suggesting that mESCs, at least partly, rely on OXPHOS for ATP production [20,21]. The discrepancies between the aforementioned studies might be explained by different degrees of OXPHOS inhibition. However, it has also been suggested that hPSCs may maintain the mitochondrial membrane potential through the ATP hydrolase activity of the dissociated F1F0 ATP synthase components, thereby ensuring their proliferation and viability [18]. Thus, the decreased ATP content observed upon mitochondrial uncoupling in PSCs might be, at least partly, due to increased ATP hydrolase activity of the F1F0 ATP synthase aimed at preserving the mitochondrial membrane potential.
Importantly, the mitochondrial ultrastructure and metabolism differ, depending on the pluripotency stage of PSCs. Two phases of pluripotency can be distinguished: naive and primed. Naive ESCs are derived from the preimplantation epiblast and display no differentiation bias unlike primed ESCs or epiblast stem cells (EpiSCs), which are derived from the postimplantation epiblast and represent a more mature stage [22]. Incidentally, recent data indicate that resetting hPSCs to a naive state (standard laboratory hESCs are similar to the mouse EpiSC stage) through transient overexpression of Nanog and KLF4 is associated with increased OXPHOS activity [23]. Furthermore, naive hESCs are less dependent on glycolysis for pluripotency maintenance than primed hESCs [23]. In mice, although EpiSCs were initially reported to be more glycolytic than mESCs, the latter displaying a bivalent metabolism [21], other studies documented that mouse EpiSCs are less glycolytic than mouse ESCs and iPSCs [24]. The use of cell lines versus primary cells may explain the discrepancies between those studies. At the morphological level, mouse EpiSCs contain more elongated mitochondria with more developed cristae than ESCs and iPSCs, although the mitochondria are still less mature than those of somatic cells [21,24]. These data emphasize that different levels of mitochondrial maturation and metabolism predilection exist, depending on the pluripotency stage. In the same vein, iPSCs maintained for extended in vitro culture periods display increased mitochondrial content and reduced potential to undergo neurogenesis in comparison with young iPSCs [25]. These data emphasize the importance of controlling the precise pluripotency stage and time in culture of stem cells in studies interested in their pluripotency and metabolic features.
Modulating Metabolism or Mitochondrial Biogenesis Interferes with Stemness, Cell Differentiation, and Reprogramming
Besides drawing simple correlations, several studies have found evidence for the direct involvement of mitochondria and metabolism in governing stemness and differentiation pathways. First, inhibiting mitochondrial function promotes pluripotency and prevents differentiation. Antimycin A, an inhibitor of mitochondrial respiratory complex III, promotes the expression of pluripotency markers, such as Nanog, while decreasing the mRNA abundance of genes related to differentiation in hESCs and mESCs [6,9,20]. Similarly, mitochondrial uncoupling by CCCP in hESCs and mESCs also increases the expression of Nanog, SOX2, and OCT4 and represses transcriptional programs necessary for embryonic lineage differentiation, such as HOX genes [6]. In addition, the ectopic expression of the uncoupling protein 2 (UCP2) impairs the metabolic shift and hPSC differentiation [18].
Second, stimulating mitochondrial biogenesis reduces pluripotency and favors cell differentiation or commitment to specific lineages. The stimulation of mitochondrial biogenesis with S-nitrosoacetylpenicillamine (SNAP) in hESCs reduces the expression of pluripotency markers [10] and enhances the differentiation of mESCs into hepatocyte-like cells [26] and mESCs and hESCs into cardiomyocytes [10,27]. The overexpression of peroxisome proliferator-activated receptor (PPAR) gamma coactivator-1α (PGC-1α), the master regulator of mitochondrial biogenesis [28], in mouse iPSCs enhances their differentiation into adipocytes [29].
Third, favoring or inhibiting the metabolic shift toward glycolysis facilitates or prevents reprogramming into iPSCs, respectively, both in human and in mouse cells. The inhibition of glycolysis using 2-deoxy-D-glucose or oxalate in somatic cells prevents reprogramming into iPSCs, whereas compounds, such as glucose, D-fructose-6-phosphate, 2,4-dinitrophenol (in response to OXPHOS uncoupling), and N-oxaloylglycine, which stimulate glycolysis, enhance reprogramming [16,19,30]. Hypoxia, which stimulates glycolysis in response to the limited availability of oxygen, necessary for OXPHOS activity, preserves the self-renewal of hESCs and prevents their spontaneous differentiation [31,32].
Mitochondrial Biogenesis and Metabolic Switches as Apparent Hallmarks of Cell Differentiation Processes
Mitochondrial biogenesis and the associated metabolic shift observed during the differentiation of PSCs appear to be hallmarks of differentiation processes, with various SSC differentiation models providing evidence for similar changes. An increased level of PGC-1α mRNA, mitochondrial mass, and mtDNA copy number, together with increased formation of OXPHOS complexes and OXPHOS activity, is observed during the adipogenic differentiation of human bone marrow-derived MSCs (hBM-MSCs) [33 –35]. Inhibiting mitochondrial biogenesis by the repression of PGC-1α [36] or mitochondrial transcription factor A (TFAM) [33] impairs adipogenic differentiation, as does rotenone-induced inhibition of mitochondrial respiration [33]. Similarly, the osteogenic differentiation of hBM-MSCs and umbilical cord-derived MSCs is accompanied by mitochondrial biogenesis, characterized by the induction of several mitochondrial biogenesis regulators, increased mtDNA copy number, cristae development, and elevated expression and activity of OXPHOS complexes [37 –39]. We recently demonstrated increases in PGC-1α expression, the mitochondria to cytoplasm ratio, mtDNA content, and OXPHOS expression and activity during the hepatogenic differentiation of hBM-MSCs [40]. In mouse HSCs (mHSCs), the mitochondrial mass increases during the early stages of differentiation [41], whereas mitochondrial respiration defects, caused by mitochondrial DNA depletion, impair their differentiation [42].
Similarities might exist in the mitochondrial content and function of normal and cancer stem cells (CSCs) as lung CSCs display reduced mitochondrial content and function compared with non-CSCs [43]. Nasopharyngeal carcinoma CSCs contain round-shaped perinuclear mitochondria similar to those observed in ESCs as well as increased glycolytic metabolism compared with their differentiated counterparts. In addition, blocking the glycolytic pathway alters the properties of CSCs [44]. These observations support the idea that relationships between mitochondria, metabolism, and stemness may underlie both physiological and pathological conditions.
Glycolysis Benefits PSCs
Glycolytic metabolism is less efficient than OXPHOS in terms of the energy production yield, raising questions as to why PSCs might favor this type of metabolism. First, a high glycolysis rate, combined with reduced OXPHOS, might be necessary for PSCs to supply the cofactors and substrates necessary for the biosynthetic reactions underlying their expansion [45]. In line with this hypothesis, the proliferative capacity of mESCs is associated with high activity of different glycolytic enzymes, elevated glycolytic flux, and low mitochondrial oxygen consumption [46]. When compared with differentiated cells, hPSCs display more phosphorylated pyruvate dehydrogenase (PDH) E1α [9], which inactivates the PDH complex and results in lower levels of substrates entering the tricarboxylic acid (TCA) cycle [47]. Similarly, the expression of pyruvate dehydrogenase kinase (PDK) 2 and 4, which prevents the mitochondrial oxidation of pyruvate through the inhibition of the PDH complex and favors glycolysis, is important for the quiescence and function of mHSCs [48]. UCP2 prevents mitochondrial glucose oxidation through a substrate-shunting mechanism, resulting in increased glycolysis and nucleotide synthesis through the pentose phosphate pathway [49]. UCP2 is highly expressed in hPSCs compared with differentiated cells. Its ectopic expression, which is normally repressed upon differentiation of PSCs, prevents any metabolic shift and impairs their differentiation [18]. PSCs also express higher mRNA levels of transketolase. Transketolase connects glycolysis to the biosynthetic pentose phosphate pathway, which is involved in nucleotide synthesis [50]. Therefore, PSCs increase the flux of glycolysis intermediates entering the pentose phosphate pathway while inhibiting their entry into the TCA cycle, thus favoring nucleotide synthesis over energy production.
The second advantage for PSCs in circumventing oxidative metabolism might be to reduce the production of ROS, which can damage genetic material and cell constituents. Maintaining low levels of ROS could be of critical importance for organismal development and homeostasis in providing protection against such damage. In contrast, increasing levels of ROS might be necessary for the commitment to specific cell types by activating particular signaling pathways, as discussed later (see The tight regulation of cell fate by ROS section).
Third, as PSCs reside in hypoxic niches, their glycolytic metabolism might represent an adaptation to their environment. In agreement with this hypothesis, hMSCs, when cultured under normoxia (21% O2), can use OXPHOS efficiently. However, expansion under 21% O2 increases senescence in MSCs when compared with physiological oxygen levels (5% O2), suggesting that a glycolysis-based metabolism favored by a hypoxic environment might protect MSCs against senescence [51].
Mitochondria and Metabolism Remodeling as Early Events in Cell Differentiation and Reprogramming Processes
Mitochondrial biogenesis and metabolic shifts are early events in multiple stem cell differentiation models, with most changes observed in the first stage of the differentiation process. Maturation of the mitochondrial network, as well as increased transcription of mtDNA, is observed during the differentiation of hESCs into cardiomyocytes [5]. The development of the mitochondrial network precedes the loss of the pluripotency markers, OCT4 and Nanog, in differentiating hESCs [6]. Mitochondrial biogenesis and metabolic shift toward OXPHOS are also early events in osteogenic [37,39], adipogenic [34], and hepatogenic [40] differentiation of hMSCs. In mHSCs, the upregulation of mitochondrial biogenesis was demonstrated to parallel the loss of pluripotency [41]. Mitochondrial respiration is more important in the commitment of mHSCs into lineage-restricted progenitor cells than in reaching the final phase of cell differentiation [42].
In contrast, the metabolic reprogramming observed during the generation of iPSCs occurs before the reestablishment of a pluripotency status [16]. Favoring glycolysis during the first days of reprogramming of human somatic cells is sufficient to increase the efficiency of the reprogramming, although no iPSCs are derived at the end of glycolysis-stimulating treatment [30]. These data suggest that enhanced glycolysis might predispose somatic cells to more efficient reprogramming into iPSCs. Consistently, the bioenergetic status of different somatic cells correlates with their reprogramming efficiencies: the closer the metabolic status of human somatic cells resembles that of PSCs, the better and faster is the reprogramming efficiency [19]. Although a glycolytic-based metabolism represents a prerequisite for efficient iPSC generation, continuous remodeling of the metabolic status of iPSCs occurs during the reprogramming time course. Mouse iPSCs more closely resemble ESCs when cultured for longer periods [52]. Similarly, in humans, the metabolome of late-passaged iPSCs (passage 41 and over) is more comparable with ESCs than that of early passaged iPSCs (passage 16) [19].
A New Shape for a New Fate: Mitochondrial Dynamics as a Regulator of Cell Fate
Mitochondrial dynamics influence most, if not all, mitochondrial-dependent biological processes, such as apoptosis, calcium homeostasis, and ATP production. Emerging evidence also points toward a role for mitochondrial fusion and fission in signaling pathways regulating stem cell proliferation and differentiation [3]. Mitochondrial elongation requires the fusion of the outer and inner mitochondrial membranes (OMM and IMM). The dynamin-related GTPases, mitofusin 1 (MFN1) and 2 (MFN2), mediate the fusion of the OMM, and the optic atrophy 1 (OPA1) protein and MFN1 mediate the fusion of the IMM. Several other proteins are involved in mitochondrial fusion, including prohibitin (PHB), which regulates OPA1 processing [53]. The cytosolic dynamin-related protein 1 (DRP1) regulates mitochondrial fission. Upon recruitment to the OMM by receptors, including the mitochondrial fission factor (MFF), the mitochondrial dynamic proteins of 49 kDa (MID49) and 51 kDa (MID51), and mitochondrial fission 1 (FIS1), DRP1 oligomerizes and induces mitochondrial constriction and fission [54]. Multiple post-translational modifications are involved in the regulation of the mitochondrial dynamic machinery [54,55].
Reduced expression 1 (REX1), a widely used pluripotency marker, was only recently found to regulate cell fate through its effect on mitochondrial dynamics [56]. Repressing REX1 expression in hPSCs results in the downregulation of pluripotency markers, upregulation of differentiation markers of all three germ layers, and development of mitochondrial morphology and activity. Mechanistically, these effects are mediated by the positive regulation of cyclin B1/B2 by REX1. This leads to increased cyclin B/cyclin-dependent kinase 1-dependent phosphorylation of DRP1 at Ser616, a post-translational modification triggering mitochondrial fission. Accordingly, the overexpression of REX1 or the DRP1-S616D mutant protein favors reprogramming into iPSCs, whereas repressing the expression of REX1 or DRP1 prevents reprogramming [56]. Although these data suggest a critical role for DRP1-mediated mitochondrial fission in the pluripotency of hESCs, studies of mESCs reported contradictory results. In mouse embryonic fibroblasts, although pharmacological inhibition of the self-assembly of DRP1 reduces the reprogramming efficiency [57], the knockdown of DRP1 expression does not affect pluripotency gene expression [58]. In addition, the increased levels of DRP1 resulting from the depletion of growth factor erv1-like, a flavin adenine dinucleotide-dependent sulfhydryl oxidase predominantly found in the intermembrane space of mitochondria, inhibit the expression of pluripotency markers in mESCs [59]. The discrepancies in the relationship of DRP1 with pluripotency might be due to species-specific responses. For example, overexpressing REX1 does not confer any advantageous effect during the reprogramming of mouse fibroblasts [56]. Although mainly considered as a component of the mitochondrial fission machinery, DRP1 can also promote mitochondrial fusion by its interaction with MFN2 [60]. The knockdown of DRP1 leads to the elongation of the mitochondrial network of fibroblasts, but it does not cause substantial changes in the morphology of mouse ESC/iPSC mitochondria [58]. These findings suggest that the discrepancies between the previous studies might also depend on DRP1 playing a different role in mitochondrial dynamics in different cells. Furthermore, tight control of DRP1 levels and/or activity, possibly regulated by different post-translational modifications, might underlie these observations. Beyond its involvement in pluripotency, DRP1-mediated fission is required for the terminal differentiation of mESCs in neuronal lineages [58] as well as for the myogenic differentiation of myoblasts [61].
Prohibitin 2 (PHB2) was recently shown to be involved in the control of stem cell fate. Overexpression of PHB2, which is normally high in mESCs, favors the proliferation of mESCs, but inhibits their differentiation into neuronal and endodermal lineages. The overexpression of PHB2 also prevents the maturation of mitochondria observed during mESC differentiation [62]. The effect of PHB2 on the maturation of mitochondria is mediated by enhanced OPA1 processing, resulting in increased mitochondrial fission and dysfunctional mitochondria [62]. The depletion of protein tyrosine phosphatase, mitochondrial 1 (PTPMT1), a mitochondrial phosphatidylinositol phosphate (PIP) phosphatase, reduces oxidative metabolism, increases glycolysis in mESCs, and prevents their differentiation. These effects are mediated by increased mitochondrial fission due to the accumulation of PIP substrates in the mitochondria [63]. Interestingly, PTPMT1 is also required for the differentiation of mHSCs. PTPMT1 acts by priming mitochondria for a rapid metabolic transition toward OXPHOS. Mechanistically, the accumulation of PIP substrates resulting from the depletion of PTPMT1 induces the activity of UCP2, which, as mentioned earlier, inhibits pyruvate oxidation in mitochondria and thus impairs metabolic shifts [64]. An increase in MFN2 and OPA1 expression is required for the differentiation of mESCs into the cardiomyocyte lineage, supporting the involvement of mitochondrial fusion in the differentiation of mESCs. Inhibiting mitochondrial fusion by repressing the expression of MFN2 and OPA1 in mESCs results in the activation of the Ca2+/calcineurin signaling pathway, leading to increased Notch activity and the repression of myocyte enhancer factor-2c and GATA-binding protein 4, two cardiomyocyte lineage transcriptional regulators [65].
Together, the aforementioned mechanistic studies demonstrate that mitochondrial dynamics, encompassing both fusion and fission events, are involved both in cell differentiation and in reprogramming events. The bidirectional influences of mitochondrial dynamics on pluripotency and differentiation are predictable because pluripotency appears to affect mitochondrial dynamics and activity, which regulates pluripotency and differentiation, as outlined in the following sections. As recently reviewed elsewhere [66], crosstalk also exists between cell cycle regulators and the components of mitochondrial dynamics. As cell cycle exit is generally required in differentiation programs, this crosstalk might also be involved in the regulation of cell differentiation.
Molecular Crosstalk Between Mitochondria, Metabolism, Pluripotency, and Differentiation
Various models have firmly established the differences in the mitochondria and metabolism of pluripotent and differentiated cells. Efforts are now underway to unveil how pluripotency and differentiation affect mitochondrial biogenesis and function and how mitochondria and metabolism modulate pluripotency and differentiation. In the following sections, we describe the pathways involved in the bidirectional crosstalk between mitochondria and metabolism and the regulation of pluripotency and differentiation (Fig. 2).
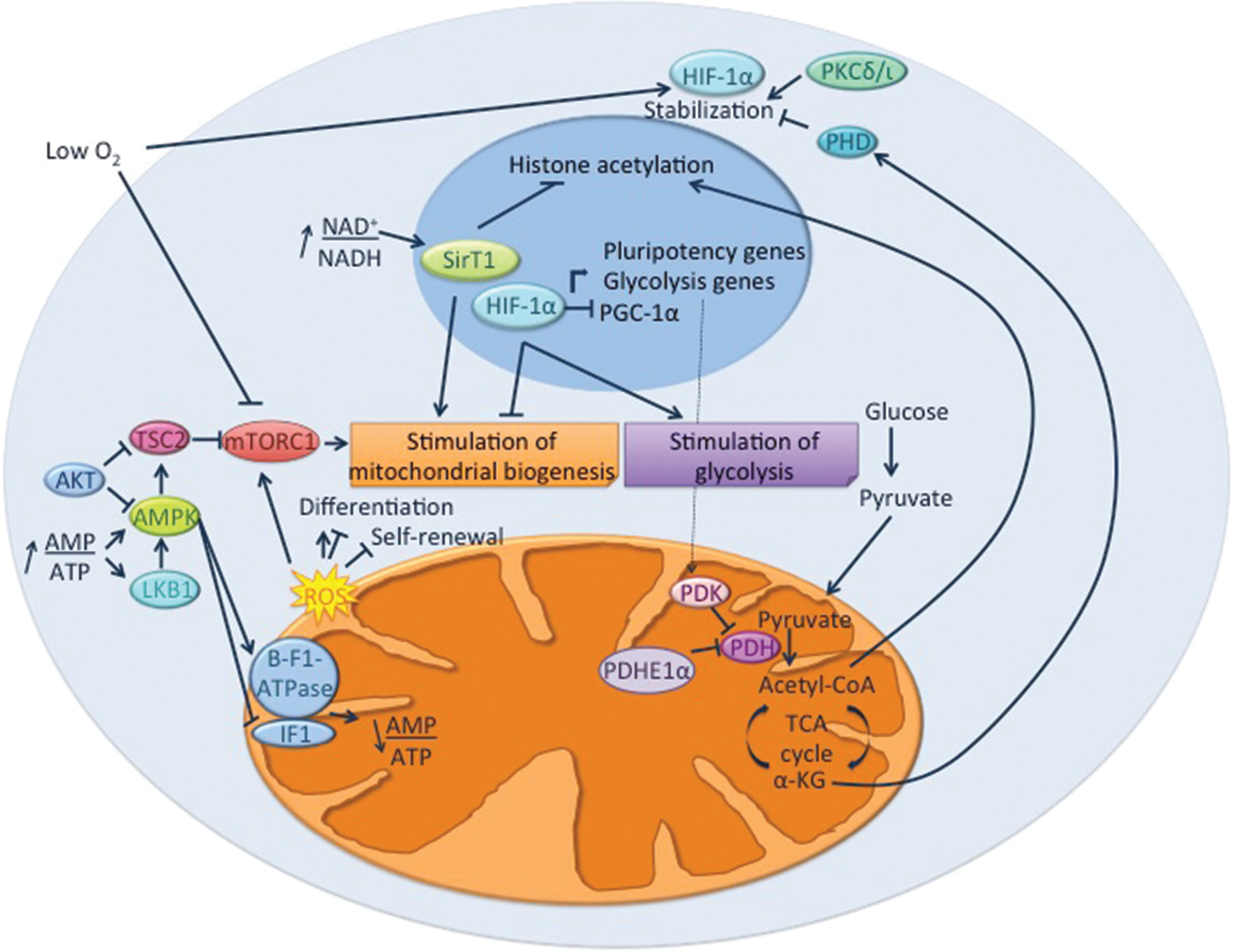
Pathways involved in the interplay between pluripotency, mitochondrial biogenesis, and metabolism. On the one hand, energy-, nutrient-, and environment-sensing pathways regulate both pluripotency and metabolism through their effects on glycolysis and mitochondrial biogenesis and activity. On the other hand, mitochondrial activity regulates stemness and differentiation through various mechanisms. These involve, for example, the production of ROS, which can induce or prevent the commitment to specific differentiation lineages; the production of intermediates or cofactors influencing epigenetic marks, protein activity, and stability; and the modification of the redox or energy status of the cells, thus altering nutrient- and energy-sensing pathways. AMPK, AMP-activated protein kinase; IF1, inhibitory factor I; LKB1, liver kinase B1; HIF-1α, hypoxia-inducible factor1α; mTORC1, mammalian target of rapamycin complex I; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1α; PHD, prolyl hydroxylase domain-containing protein; PKCλ/ι, protein kinase C isoform lambda/iota; TCA, tricarboxilic acid; TSC2, tuberous sclerosis complex 2; α-KG, α-ketoglutarate. Color images available online at
Oxygen sensing and the HIF pathway
Oxygen sensing plays a central role in metabolic homeostasis and is partly regulated by the hypoxia-inducible factor (HIF) family of transcription factors. HIF-1 is a heterodimer containing a constitutively expressed β subunit as well as an oxygen-regulated α subunit that is continuously synthesized and then degraded by prolyl hydroxylases under normoxia. In response to hypoxia, HIF-1 activation promotes a switch from mitochondrial respiration to anaerobic glycolysis and suppresses mitochondrial biogenesis [67]. Interestingly, most stem cell types reside in hypoxic niches, and increasing evidence points toward a critical role for oxygen in the regulation of the function of stem cells [68]. Thus, it is not surprising that several lines of evidence support a role for HIF-1 in the interplay between mitochondria, metabolism, and stem cell functions. As HIF-1 is involved in the dedifferentiation of cancer cells [69], it might also regulate differentiation pathways in pathological conditions.
The protein kinase C isoform lambda/iota (PKCλ/ι) is involved in the coregulation of mitochondrial biogenesis and stem cell pluripotency/differentiation through the HIF-1α pathway [70]. The depletion of PKCλ/ι induces a metabolic shift, resulting in increased glycolysis and decreased OXPHOS activity. It also promotes naive pluripotency in mESCs. These effects are mediated by stabilization of HIF-1α, which positively regulates the expression of pluripotency genes while suppressing the expression of PGC-1α by direct binding to their promoters. Thus, HIF-1α simultaneously regulates pluripotency and mitochondrial biogenesis by activating and repressing transcriptional activities, respectively [70].
The involvement of HIF-1α in pluripotency is not limited to ESCs. Upon reprogramming of human cells, HIF-1α improves the induction of pluripotency by activating a glycolytic program, predisposing the somatic cells to more efficient reprogramming [71]. The expression of HIF-1α is also important for the quiescence and function of mHSCs. It maintains the expression of PDK, which, by inactivating the PDH complex, prevents the mitochondrial oxidation of acetyl-CoA and favors glycolysis. On the other hand, HIF-1α deficiency results in a shift from glycolysis to OXPHOS and is associated with increased mitochondrial mass in HSCs [48]. In hMSCs, hypoxia or the stabilization of HIF-1α using cobalt chloride prevents the osteogenic differentiation and the associated increase in mitochondrial biogenesis [72].
Thus, HIF-1α can control pluripotency gene expression and promote glycolytic metabolism while inhibiting mitochondrial biogenesis and function. It remains to be determined whether PKCλ/ι acts systematically upstream of HIF-1α. Other questions that need to be addressed are whether other upstream regulators are involved in the activation of HIF-1α and whether such activation depends on cell types or lineage commitment. In addition, the roles of other molecular actors (ie, whether they act together or independently of HIF-1α) in the control of pluripotency gene expression and activation of glycolysis need to be determined.
The tight regulation of cell fate by ROS
ROS are highly reactive reduced forms of molecular oxygen, such as the superoxide radical anion (O2•−) and hydrogen peroxide H2O2. Under physiological conditions, ROS are naturally produced by the respiratory chain during OXPHOS [73]. Although ROS exert oxidative damage on lipids, proteins, and DNA, they also act as secondary messengers and were recently shown to be involved in the regulation of stem cell self-renewal, pluripotency, and differentiation [74].
Although a specific role for mitochondrial ROS has yet to be demonstrated in ESCs, stimulating or inhibiting the production of ROS favors or prevents mESC differentiation into cardiomyocytes, respectively [75 –79]. However, it needs to be emphasized that the stimulating effect of ROS on cardiomyocyte differentiation might strongly depend on the level and nature of the ROS. Indeed, mESCs simultaneously treated with antioxidants and cyclosporin A, an inhibitor of the mitochondrial permeability transition pore (mPTP) resulting in increased mitochondrial biogenesis, activity, and ROS generation, synergistically favor cardiomyocyte differentiation [80].
ROS regulate the proliferation and differentiation of rat neural progenitors, with proliferating neural progenitors displaying low levels of ROS in comparison with their differentiated neuronal and astrocytic counterparts [81]. A recent study showed that rapid bursts of superoxide radical anions called superoxide flashes, which are highly reactive reduced forms of molecular oxygen, were involved in regulating the self-renewal and differentiation of mouse embryonic neural progenitor cells (NPCs). Mitochondrial superoxide scavengers or mPTP inhibitors reduce the frequency of superoxide flashes and enhance NPC proliferation [82]. In contrast, increased superoxide flashes are necessary for the differentiation of mouse NPCs into cortical neurons, and inhibiting mPTP or scavenging mitochondrial ROS impairs differentiation [83]. In a mouse model of adult hippocampal neurogenesis, a peak in mitochondrial abundance and ROS levels was observed in a highly proliferative, intermediate progenitor state, but not in undifferentiated neural stem cells or postmitotic neurons [84]. Although these differences might depend on the embryonic or adult status of the NPCs, the data suggest that the involvement of ROS in neurogenesis might be limited only to a specific stage of cell differentiation.
In hMSCs, mitochondrial complex III-derived ROS, independently of OXPHOS, are required for the expression of PPARγ and initiation of the adipogenic transcriptional program [34]. Mitochondria-targeted antioxidants such as mitoCP prevent both the adipogenic differentiation and the associated increase in ROS, whereas the addition of exogenous H2O2 to mitoCP-treated hMSCs rescues adipogenic differentiation [34]. Similarly, hypoxia stimulates adipocyte differentiation by enhancing the production of mitochondrial ROS [85]. On the other hand, both exogenous H2O2 and oligomycin-dependent inhibition of mitochondrial activity inhibit the osteogenic differentiation of hMSCs. These data suggest that mitochondrial OXPHOS is required for osteogenic differentiation and that excessive ROS hamper osteogenic differentiation [37]. Beyond their role in MSC differentiation, mitochondrial ROS are also involved in MSC senescence. The treatment of mouse MSCs with transforming growth factor β1 induces their senescence. This effect is, at least partly, mediated by the generation of mitochondrial ROS, suggesting that maintaining low levels of mitochondrial ROS is necessary to preserve MSC stemness [86].
The involvement of ROS in the function of HSCs has also been established and recently reviewed [74,87]. mHSCs with low ROS levels are more quiescent and display increased self-renewal potential compared with HSCs displaying higher ROS levels, which are prone to exhaustion upon serial transplantations [88]. The negative impact of ROS on mHSC self-renewal is mediated by the activation of p38 MAPK and mammalian target of rapamycin (mTOR) pathways, with studies showing that inhibiting either of these pathways restores the long-term reconstitution ability of HSCs with high levels of ROS [88,89]. In contrast, ROS triggered the differentiation of a Drosophila population of hematopoietic progenitors resembling mammalian myeloid progenitors through the activation of c-Jun N-terminal kinases, leading to activation of Forkhead box O and inhibition of the activity of polycomb [90]. Similarly, increasing ROS in AKT1/AKT2 double-deficient mHSCs, which show lower ROS levels, rescues the defects in differentiation [91]. Thus, low levels of ROS are necessary to preserve the quiescence of HSCs, and increased ROS levels favor HSC differentiation.
The generation of mitochondrial ROS may also represent a key upstream signaling event in epidermal differentiation and hair follicle development [92]. In mouse keratinocytes, preventing the generation of mitochondrial ROS through the ablation of TFAM, which is required for the transcription of mtDNA-encoded mitochondrial genes [93], disturbs the transduction of Notch signaling required for keratinocyte differentiation [92].
Based on the aforementioned, maintaining low ROS levels, notably through reduced mitochondrial activity, appears necessary to preserve the self-renewal ability of different types of stem cells, and increasing ROS seems to favor particular lineage commitment during differentiation. Maintaining low ROS levels in stem cells, possibly through reduced mitochondrial function, might represent a protective mechanism of the genome against ROS-mediated damage, which is essential to ensure optimal development and organism homeostasis upon aging. Further studies are required to elucidate more precisely which type(s) of ROS are endogenously produced in different models of stem cell differentiation and whether different types of ROS exert similar or opposite effects on pluripotency and differentiation in various models.
Nutrient- and energy-sensing pathways: a complex connection between energy metabolism and stem cell function
Given that stem cells and differentiated cells display different metabolisms and metabolomes, it is not surprising that nutrient- and energy-sensing pathways are involved in the regulation of stem cell pluripotency and differentiation. The following sections illustrate the roles of major sensing pathways in orchestrating the interplay between mitochondria, metabolism, stem cell pluripotency, and differentiation.
The mTOR pathway
The mTOR pathway plays a crucial regulatory role in cell metabolism, proliferation, growth, and survival by sensing extracellular and intracellular signals, such as oxygen, and the energy status, growth factors, and amino acid content. Two distinct complexes, mTORC1 and mTORC2, exist and exert overlapping but distinct functions. The mTORC1 complex is sensitive to amino acids, stress, oxygen, energy, and growth factors. It promotes cell growth and cell cycle progression by inducing anabolic processes, such as the biosynthesis of proteins and lipids and the stimulation of mitochondrial biogenesis and metabolism, while limiting catabolic processes, including autophagy. The mTORC2 complex, which has not been described in as much detail as the mTORC1 complex, responds to growth factors and regulates metabolism, cell survival, and cytoskeletal organization [94,95]. The mTORC1 complex is acutely sensitive to rapamycin treatment [95]. In contrast, the mTORC2 complex is insensitive to acute rapamycin treatment, although chronic exposure disrupts the mTORC2 pathway [95]. The mTORC1 pathway is involved in the regulation of pluripotency and differentiation, notably through its relationship with mitochondrial biogenesis.
The involvement of mTORC1 in ESC pluripotency and differentiation appears complex. On the one hand, mTOR activity is required for the proliferation of hESCs and mESCs [96] and its inhibition impairs pluripotency in hESCs [97]. The sorting of mESCs according to low or high mitochondrial membrane potential also reflects the activation status of mTORC1 in ESCs and is correlated with pluripotency and differentiation capacities [98]. Treatment with rapamycin decreases pluripotency and increases the differentiation capacities of mESCs with high mitochondrial membrane potential [98]. On the other hand, Deptor, an endogenous inhibitor of mTORC1 and mTORC2 activity, maintains mESC pluripotency, and repression of Deptor triggers differentiation [99]. Increased mTORC1 activity has been consistently shown during the differentiation of mESCs and hESCs into a neuronal lineage [99,100] and the differentiation of hESCs into fibroblast-like cells [101]. In mice, rapamycin-induced inhibition of mTOR activity increases the reprogramming efficiency [102], whereas the overactivation of the mTOR pathway in cells lacking tuberous sclerosis complex 2, a negative regulator of mTORC1 [95], impairs reprogramming [103]. Likely, tight control of mTOR signaling determines the outcome of pluripotency and differentiation, possibly with differences related to lineages, species, and the relative contribution of mTORC1 and mTORC2 signaling. In agreement with this hypothesis, treatment of hESCs with 100 nM rapamycin for 6 days results in a loss of the pluripotency markers, OCT-4, Nanog, and SOX2 [97], whereas treatment with 20 nM rapamycin for 3 days has no impact on pluripotency gene expression [101]. The differences in mTOR activity may also be due to the precise stage of pluripotency (naive vs. primed ESCs, or ESCs vs. epiSCs), with naive ESCs having increased mTOR activity and mitochondrial biogenesis and function compared with primed ESCs/epiSCs.
The involvement of mTOR has also been studied in SSCs. Reduced activity of mTORC1 signaling preserves mHSC self-renewal and differentiation potential by repressing mitochondrial biogenesis and ROS production [104]. Nonetheless, mTORC1 activity is still required for mHSC function as the deletion of the regulatory-associated protein of mTOR (Raptor) ablates mTORC1 activity and results in defective hematopoiesis [105]. There also seems to be a link between mTORC1 signaling and stem cell function, partly dependent on ROS, in hMSCs. During the adipocyte differentiation of hMSCs, mTORC1 is required for the generation of ROS, which is also required for the differentiation process (see The tight regulation of cell fate by ROS section). Although overexpression of PPARγ facilitates adipogenesis in the presence of antioxidants, this effect is lost upon PI3K or mTORC1 inhibition, suggesting that mTORC1 additionally favors adipocyte differentiation by effectors other than ROS [34].
Collectively, these data suggest that the magnitude and duration of mTORC1 activation or inhibition, rather than an on/off status, likely influence stem cell pluripotency and differentiation. Different effectors downstream of mTORC1 signaling might be involved, depending on the stem cell type and differentiation lineages. Most studies have focused on mTORC1. The contribution of mTORC2 remains elusive and should be the focus of further experiments. Given the central role of metabolism in regulating stemness and differentiation, it is important to investigate the effectors that connect the activity status of mTOR with stemness and differentiation.
AMPK and its upstream regulators
AMP-activated protein kinase (AMPK) is a key heterotrimeric, intracellular energy sensor sensitive to the AMP/ATP ratio. In low-energy conditions (a high AMP/ATP ratio), AMPK is activated and induces catabolic pathways, such as fatty acid oxidation or glucose uptake, while inhibiting anabolic pathways, including lipid and protein synthesis, to restore the energy balance [106].
Downregulated expression of the catalytic α1 subunit of AMPK is found in hESCs and iPSCs when compared with differentiated cells [107]. Interestingly, metformin-activated AMPK impedes human and mouse iPSC generation through the establishment of a metabolic barrier distinct from the enhanced glycolysis required for reprogramming [108]. Mechanistically, the activation of AMPK upregulates the catalytic β-F1-ATPase subunit, a rate-limiting component of mitochondrial OXPHOS normally repressed upon reprogramming. Simultaneously, AMPK activation prevents the induction of the ATPase inhibitory factor 1, thus impeding the metabolic shift required for reprogramming [109]. However, the role of AMPK in pluripotency appears very complex as the activation of AMPK in mESCs is associated with upregulation of pluripotency genes [110]. Thus, AMPK activity is likely not directly involved in the promotion of pluripotent versus differentiated cells. Instead, the intracellular environment, comprising a precise metabolome and proteome at a given moment, as well as the activation of different effectors, likely influences the outcome of AMPK activity in determining the fate of stem cells. Accordingly, the involvement of AMPK in cell differentiation differs, depending on the lineage commitment. AMPK activity inhibits mouse myoblast differentiation [111], whereas it favors human endothelial [112] and mouse cardiac [113] differentiation. In the latter model, the simultaneous knockdown of several adenylate kinase isoforms normally induced during cardiogenesis decreases AMPK activity and impedes cardiomyocyte differentiation and associated mitochondrial biogenesis [113]. The production of nitric oxide, a key regulator of cardiomyogenesis in mESCs and potent inducer of mitochondrial biogenesis, occurs downstream of AMPK and mTORC1 signaling pathways [114]. Thus, these two pathways may interact to control the interplay between mitochondria, metabolism, stemness, and differentiation.
AMPK is also involved in the regulation of SSC pluripotency and differentiation. As recently reviewed elsewhere, AMPK activity controls the fate of MSCs by favoring osteogenic and inhibiting adipogenic differentiation. The role of AMPK in controlling the differentiation of MSCs may depend on its involvement in multiple pathways, including the ERK pathway, mTOR signaling, Wnt/β-catenin signaling, and energy metabolism [115]. In mHSCs, the constitutively active liver kinase B1 (LKB1), which activates AMPK and AMPK-related kinases (interested readers can refer to [116]) under low-energy conditions, is essential to preserve HSC quiescence [117 –119]. The effects of LKB1 on mHSC quiescence might involve the protection of their mitochondria. Indeed, LKB1-deficient mHSCs have reduced mtDNA content, mitochondrial membrane potential, PGC-1α and -β levels, and ATP levels, despite increased mitochondrial mass. These observations suggest that mitochondrial dysfunction occurs in LKB1-deficient mHSCs [117,118]. Further studies are required to determine whether this mitochondrial dysfunction is a direct consequence of LKB1 inactivation. Examining whether mitochondrial dysfunction plays a direct or indirect role in the disruption of HSC quiescence in LKB1-deficient HSCs is also important.
Metabolites and cofactors: small molecules with a broad influence
Due to their reliance on different types of metabolisms, PSCs and differentiated cells display distinct metabolomes. A number of metabolites directly modulate gene expression by regulating the activity/stability of transcription factors and by inducing epigenetic modifications [120]. Global chromatin remodeling, characterized by a transition from relatively open chromatin to more compact chromatin, occurs during cell differentiation. Evidence supports a direct role for epigenetic modifications in driving cell fate decisions rather than stabilizing gene expression [121]. In the following sections, we propose several ways in which the differences in metabolomes could affect gene expression and control cell differentiation.
The redox state and Sirtuin proteins
The NAD+/NADH ratio, which is dependent on glycolytic and mitochondrial metabolisms, might regulate the cell fate by modulating NAD+-dependent deacetylase enzymes, such as sirtuins. These proteins are involved in the control of gene expression and epigenetics through deacetylation of transcriptional regulators or histone proteins [122]. SIRT1 levels are decreased during hESC and mESC differentiation [123,124]. Although SIRT1 is not required for mHSC function [125], some reports suggest that SIRT1 might be required for both the maintenance and differentiation of mHSCs [126]. In MSCs, SIRT1 activity favors osteogenesis while inhibiting adipogenesis [115]. The accumulated evidence clearly supports the involvement of SIRT1 in the regulation of pluripotency and differentiation processes, although the mechanisms underlying stage and lineage specificities remain obscure. Bidirectional crosstalk between the activity of sirtuins and mitochondrial metabolism might exist as sirtuins can induce mitochondrial biogenesis [127] and regulate mitochondrial energy metabolism through the post-translational regulation of mitochondrial proteins [128]. Therefore, further studies should assess the putative bidirectional influences of SIRT1 and mitochondrial biogenesis and function in various models of stem cell differentiation.
TCA cycle metabolites: roles inside and outside the mitochondria
Intermediates of the TCA cycle might also be involved in determining cell fate. For example, the α-ketoglutarate metabolite can exit mitochondria and promote DNA and histone demethylation through the activation of 10–11 translocation enzymes, lysine demethylases, and Jumonji C domain-containing demethylases. α-Ketoglutarate is also involved in the stability of HIF-1 through the activation of the prolyl hydroxylase domain-containing protein 2, stimulating the degradation of HIF-1α [120]. This might be of critical significance in the interplay between mitochondria, metabolism, and pluripotency, given the recently discovered role of HIF-1α in the coregulation of those processes [70]. Similarly, the acetyl-coenzyme A cofactor, which is mainly produced downstream of glycolysis and fatty acid oxidation, can, instead of feeding the TCA cycle, exit mitochondria and reach the nucleus where it promotes histone acetylation, as described in a model of adipocyte differentiation [129].
Noncoding RNAs: a new way to decode the interplay between mitochondria and cell differentiation?
The role of noncoding RNAs, such as microRNAs (miRNAs) or long noncoding RNAs (lncRNAs) in many biological processes, including stem cell differentiation and metabolism regulation, is increasingly being appreciated. Their broad impact, resulting from their various mechanisms of action, as well as high number of targets, a makes noncoding RNAs appealing candidates for the coregulation of simultaneous processes. On the one hand, miR-1, a myomir induced during myogenic differentiation, was reported to inhibit the expression of a transcriptional repressor of muscle gene expression [130], but on the other hand, it was reported to enter the mitochondria and stimulate the translation of specific mitochondrial genome-encoded transcripts [131]. These data suggest that miR-1 might coordinate the myogenic program by coregulating mitochondrial biogenesis and the myogenic-associated transcriptional program by distinct mechanisms [131]. In a recent study, a single-cell transcriptome analysis of several cell types undergoing reprogramming into iPSCs identified two lncRNAs involved in the regulation of multiple mitochondrial and metabolic targets [132]. A number of noncoding RNAs have reported roles in regulating cell fate or cell metabolism and mitochondrial biogenesis. Future studies should assess the relevance of those candidates as putative global coregulators of cellular fate and mitochondrial biogenesis and function.
Conclusion and Perspectives
The deep remodeling of mitochondria observed during stem cell differentiation and reprogramming has unveiled a critical role for glycolytic and oxidative metabolisms in the regulation of self-renewal, pluripotency, plasticity, and differentiation of stem cells. PSCs rely heavily on glycolysis, which provides rapid energy production and the substrates necessary for proliferation, is adapted to a hypoxic niche, and enables limited production of ROS. On the other hand, differentiated cells display a more developed and functional mitochondrial network and rely heavily on OXPHOS. Accordingly, mitochondrial function directly regulates stem cell differentiation through various mechanisms involving ROS production, metabolomic modifications, and modulation of the redox and energy status. Many of the pathways involved in the interplay between glycolytic and oxidative metabolisms on the one hand, and pluripotency on the other hand, act bidirectionally and interact with each other. This leads to a complex and still unresolved question: Is the mitochondrial biogenesis initiated first, followed by a loss of pluripotency and a commitment to differentiation, which is further accompanied by increased mitochondrial function? Or does the loss of pluripotency initiate mitochondrial biogenesis and a metabolic shift? Single cell tracking and combined OMICS technologies might help to resolve this question, as well as to further elucidate the underlying pathways. It would be intriguing to study how mitochondria are distributed and how they evolve in self-renewing or committed cells upon asymmetric division. Efforts should be made to study mitochondrial and metabolic changes upon in vivo differentiation. The recent development of genetically encoded fluorescent probes, such as roGFPmito [133], which can be used to assess the mitochondrial oxidation state, or mitoTimer [134], which can be used to follow mitochondrial protein turnover/segregation, will likely help to address these questions. Given the considerable therapeutic potential of stem cell therapy, improving our knowledge about the mechanisms regulating their self-renewal and differentiation, which may involve mitochondria, is fundamental.
Footnotes
Acknowledgment
The authors thank the Association Belge contre les Maladies Neuro-Musculaires (ABMM, Belgium) for their support.
Author Disclosure Statement
The authors declare no conflicts of interest.