
Abstract
The lack of robust Parkinson's disease (PD) phenotype in parkin knockout rodents and the identification of defective dopaminergic (DA) neurotransmission in midbrain DA neurons derived from induced pluripotent stem cells (iPSC) of PD patients with parkin mutations demonstrate the utility of patient-specific iPSCs as an effective system to model the unique vulnerabilities of midbrain DA neurons in PD. Significant efforts have been directed at developing efficient genomic engineering technologies in human iPSCs to study diseases such as PD. In the present study, we converted patient-specific iPSCs from the primed state to a naivetropic state by DOX-induced expression of transgenes (Oct4, Sox2, Klf4, c-Myc, and Nanog) and the use of 2iL (MEK inhibitor PD0325901, GSK3 inhibitor CHIR99021, and human LIF). These patient-specific naivetropic iPSCs were pluripotent in terms of marker expression, spontaneous differentiation in vitro, and teratoma formation in vivo. They exhibited morphological, proliferative, and clonogenic characteristics very similar to naive mouse embryonic stem cells (ESC). The high clonal efficiency and proliferation rate of naivetropic iPSCs enabled very efficient gene targeting of GFP to the PITX3 locus by transcription activator-like effector nuclease. The naivetropic iPSCs could be readily reverted to the primed state upon the withdrawal of DOX, 2iL, and the switch to primed-state hESC culture conditions. Midbrain DA neurons differentiated from the reverted iPSCs retained the original phenotypes caused by parkin mutations, attesting to the robustness of these phenotypes and the usefulness of genomic engineering in patient-specific naivetropic iPSCs for studying PD.
Introduction
P
Genetic modification in animal models has generated significant information on the functions of a particular gene in an organism. The ability to do the same thing in human iPSCs would greatly improve our ability to understand how mutations of genes such as parkin cause PD. There had been great difficulties to perform gene targeting in human pluripotent stem cells (PSC), because of their very low clonogenicity and the inefficiency of homologous recombination. The first issue appears to be an attribute of primed pluripotency, as human PSCs are similar to mouse epiblast stem cells [13,14], unlike mouse embryonic stem cells (ESCs), which are in an earlier, naive state of pluripotency that resembles the state of naive epiblasts in mature blastocysts [15]. There have been intense efforts to convert human PSCs from the primed state to the naive state for a fundamental understanding of pluripotency and a variety of applications, including gene targeting. Initial successes rely on the expression of pluripotency transgenes and the 2iL (MEK inhibitor PD0325901, GSK3 inhibitor CHIR99021, and human LIF) culture condition [16 –19], as 2iL supports highly efficient derivation of naive ESCs from virtually any strains of mice [20 –22]. More recently, transgene-independent naive human PSCs have been derived using a variety of small-molecule compounds in addition to 2iL [23 –27].
Engineered site-specific DNA nucleases such as zinc finger nuclease [28], transcription activator-like effector nuclease (TALEN) [29], and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) [30] markedly increase the rate of homologous recombination by introducing double-stranded DNA breaks. Recent studies have demonstrated that the use of TALEN or CRISPR in primed-state human PSC does not produce any significant off-target effect [31 –33], thus paving the way for wide adoption of these powerful technologies in modeling human diseases. However, gene targeting in primed-state human PSCs does not realize the full potentials of TALEN and CRISPER in improving homologous recombination, as primed-state human PSCs have very low clonal efficiency and slow growth rate.
In the present study, we take advantage of the patient-specific primed iPSCs that we have generated previously using DOX-inducible lentiviruses expressing Oct4, Sox2, Klf4, c-Myc, and Nanog [10]. Using DOX and 2iL, we converted these primed iPSCs to a state of pluripotency that shared many important functional similarities to naive pluripotency in mouse ESCs. We named it naivetropic state as the expression of transgenes was needed to maintain the state [16 –19], in contrast to the transgene-independent naive state in mouse [20 –22] and human [23 –27]. The naivetropic iPSCs maintained pluripotency and exhibited growth rate and clonal efficiency very similar to those of naive mouse ESCs. TALEN-mediated gene targeting of eGFP to the PITX3 locus was significantly more efficient than that achieved in primed human PSCs using the same targeting construct and TALEN pairs [29]. Midbrain DA neurons differentiated from the naivetropic iPSCs retained the dopamine-specific phenotypes found in the parental primed iPSCs. These results attest to the robustness of these phenotypes and pave the way for future experiments using genetically engineered naivetropic iPSCs to study PD in terms of its molecular etiology, cellular mechanism, and therapeutic development.
Materials and Methods
Culture of human primed iPSCs
Under the approval of the Health Sciences Institutional Review Board in the State University of New York at Buffalo, human primed iPSC lines C001, C002, P001, and P002 [10] were maintained on mitomycin C-treated mouse embryonic fibroblast (MEF) feeder cells in hESC medium (DMEM/F12 containing 20% knockout serum replacement, 2 mM
Derivation and maintenance of naivetropic iPSCs
Human primed iPSCs were treated with 10 μM Rho-associated kinase (ROCK) inhibitor Y27632 (Abcam) overnight and then trypsinized into single cells with 0.25% trypsin/EDTA (Life Technologies) for 10 min at 37°C. The single cells were plated on MEF feeders (5–6 × 105/cm2) in naivetropic iPSC medium (50% DMEM/F12 and 50% Neurobasal, with N2, B27, 1 mM glutamine, 1% NEAA, 0.1 mM β-mercaptoethanol, penicillin–streptomycin (all from Life Technologies), 20 ng/mL recombinant human LIF (Millipore), 5 mg/mL bovine serum albumin (BSA) (Sigma), 2 μg/mL doxycycline (Sigma), 1 μM PD0325901 (EMD Millipore), and 3 μM CHIR99021 (Stemgent) and cultured for 7–10 days. The small, bright, compact cell clumps in the culture were picked manually and trypsinized into single cells, which were replated on fresh MEF feeders. After several passages, mouse ESC-like dome-shaped colonies were trypsinized to single cells and passaged every 2–3 days. Naivetropic iPSCs were derived and cultured in an incubator with 5% O2.
Reversion of naivetropic iPSCs to the primed state
Naivetropic iPSC colonies were picked manually and washed one time in DMEM/F12 medium before being seeded on MEF feeder in hESC medium with bFGF. Typical iPSC colonies (designated as reverted iPSC, riPSC) appeared 7–10 days later. The riPSCs were passaged every 5–7 days using dispase (1 mg/mL). All primed-state PSCs, including riPSCs were cultured in 21% O2.
Immunostaining
Cells were cultured on gelatin- or MEF-coated glass coverslips in 24-well plate. They were fixed in 4% paraformaldehyde (Sigma) for 20 min, permeabilized with 0.1% Triton X-100 for 15 min at room temperature (RT), blocked in 3% BSA for 30 min at RT, and then incubated in primary antibody overnight at 4°C and secondary antibody for 1 h at 37°C. Primary antibodies (all from Millipore) used in this study are SSEA-1 (1:1,000), SSEA-3 (1:1,000), SSEA-4 (1:1,000), TRA-1-60 (1:1,000), TRA-1-81 (1:1,000), Oct4 (1:1,000), Nanog (1:500). Secondary antibodies were AlexaFluor 488 and 594 (1:1,000, Invitrogen). Alkaline phosphatase (AP) activity was detected by the Alkaline Phosphatase Kit (Millipore).
Measurement of cell doubling time
We plated 1 × 104 naivetropic iPSCs on gelatin-coated 24-well plates. The numbers of cells from three wells were counted with Trypan blue and averaged at days 3, 6, 9, and 12 after plating. We also plated 1 × 104 primed iPSCs on MEF feeders in 24-well plates and counted the cells in the same way at days 6 and 12 after plating. Cell doubling time was calculated using a web-based calculator (
Teratoma formation assays
The assay was performed by the Mouse Tumor Model Resource at Roswell Park Cancer Institute (Buffalo, NY) following approved IACUC protocol. Briefly, 1 million naivetropic iPSCs cultured on gelatin were mixed with collagen at a 1:1 volume ratio. About 10 μL of this mixture were placed on parafilm to solidify for 1 h. Three cell/collagen pellets were grafted under the renal capsule of each kidney in a SCID mouse (C.B-Igh-1bIcrTac-Prkdcscid/Ros). Animals were monitored for palpable tumors around the kidney area. Large tumors (∼1 cm in size) were found for each iPSC line 2–3 months after grafting. Tumors were harvested and dissected into small pieces and fixed for 24 h in 10% formalin and processed for paraffin embedding. Tissue sections (5 μm) were stained with Hematoxylin and eosin for histological identification.
Directed differentiation of reverted iPSCs to midbrain DA neurons
The differentiation was performed according to a rosette-based protocol published previously [10]. Reverted iPSCs were dissociated and grown as embryoid bodies (EB) in hESC medium with 10 μM SB431542 (Tocris Bioscience) for 4 days. The EBs were cultured in suspension in Neural Induction Media (DMEM/F12 with 1 × N2 supplements, 0.1 mM NEAA, and 2 μg/mL heparin) containing 20 ng/mL bFGF for two more days. Then the EBs were plated on Matrigel-coated six-well plates in Neural Induction Media for a week. Cells in the center of many colonies formed rosette-like structures. These primitive neuroepithelial cells were further differentiated to definitive neuroepithelia in Neural Induction Media with 20 ng/mL FGF8a (PeproTech) and 100 ng/mL SHH (PeproTech) for one more week. Rosettes were isolated manually and cultured in suspension to form neurospheres, which were cultured in Neural Induction Media with 50 ng/mL FGF8a, 100 ng/mL SHH, B27, and ascorbic acid (200 μM) for 6 days to be patterned to a mid/hind brain destiny. Neurospheres were dissociated with accutase/trypsin (1:1) to single cells, which were plated at 2 × 105 cells/mL on six-well plates or coverslips precoated with Poly-L-ornithine/laminin/Matrigel in Neural Differentiation Medium (Neurobasal medium, N2, B27, NEAA, 1 μg/mL laminin, 1 μM cAMP, 50 ng/mL FGF8a, 100 ng/mL SHH, 200 μM ascorbic acid, 1 ng/mL TGFβ3, 10 ng/mL BDGF, and 10 ng/mL GDNF). Media were half changed every other day. FGF8a was withdrawn 7 days later, SHH 2 weeks later. All differentiation experiments were conducted in 21% O2.
Measurement of dopamine release
Dopamine release was measured as described previously [10]. Briefly, iPSC-derived midbrain neurons in six-well plates were treated at 37°C in three wells (a, b, c) for 30 min. Well (a) was incubated with 1 mL hank's balanced salt solution (HBSS); well (b) with 1 mL HBSS for 15 min and then 56 mM KCl for another 15 min; well (c) with 1 mL HBSS without Ca2+ and without Mg2+, but with 2 mM EDTA for 15 min and then 56 mM KCl for another 15 min. The 1 mL solutions were taken from the wells and added with GSH and EGTA to 2 mM to minimize dopamine oxidation. Dopamine in the solutions was measured by reversed phase HPLC (ESA Model 582 with ESA MD150 × 3.2 column, at 0.6 mL/min flow rate in MD-TM mobile phase) coupled with electrochemical detection [34] (ESA Coulochem III, E1: −250 mV, 2 μA; E2: 350 mV, 2 μA). Cells in the three wells were lysed in 0.5 N NaOH to measure protein levels, which were used to normalize DA release. Spontaneous dopamine release is reflected in (a), while Ca2+- and activity-dependent release is reflected in (b and c).
Measurement of specific dopamine uptake
Dopamine uptake was measured as described previously [10]. Briefly, iPSC-derived midbrain neurons in six-well plates were washed with 1 mL prewarmed uptake buffer (10 mM HEPES, 130 mM NaCl, 1.3 mM KCl, 2.2 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 10 mM glucose, pH 7.4) three times. Cells were incubated for 10 min at 37°C with 1 mL uptake buffer containing 5 μM dopamine without or with 10 μM nomifensine (a selective inhibitor of dopamine transporter). After the cells were washed at least three times in the uptake buffer, they were lysed in 0.1 M perchloric acid with 1 mM EDTA and 0.1 mM sodium bisulfite. Cleared cell lysates were analyzed for dopamine on HPLC coupled with electrochemical detection [34] (E1: −150 mV, 2 μA; E2: 220 mV, 2 μA). The pellets were dissolved in 1 mL 0.5N NaOH to measure protein contents, which were used to normalize dopamine uptake. Specific dopamine uptake is reflected in the difference of DA content in the absence and presence of the DAT inhibitor nomifensine. The amount of dopamine in the iPSC-derived midbrain neurons without any treatment was also measured.
Electrophysiology
Recordings of synaptic and ionic currents used standard whole-cell voltage-clamp techniques. Recordings were obtained with Axopatch 200B or Axopatch 700B amplifiers that were controlled and monitored with a computer running pCLAMP (version 9) with DigiData 1320 series interfaces (Axon Instruments). Electrode resistances were typically 2–4 MΩ in the bath. After seal rupture, series resistance (4–10MΩ) was compensated (70%–90%) and periodically monitored. For the recording of spontaneous excitatory postsynaptic currents (sEPSC), membrane potential was held at −70 mV. The external solution artificial cerebro-spinal fluid (ACSF) contained (in mM): 130 NaCl, 3 KCl, 5 MgCl2, 1 CaCl2, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, pH 7.3–7.4, 300–305 mOsm/L. To isolate AMPAR-mediated response, the NMDA receptor antagonist D-aminophosphonovalerate (APV, 20 μM) and GABAA receptor antagonist bicuculline (10 μM) were added. The internal solution consisted of the following (in mM): 130 Cs methanesulfonate, 10 CsCl, 4 NaCl, 10 HEPES, 1 MgCl2, 5 EGTA, 2.2 QX-314, 12 phosphocreatine, 5 MgATP, 0.5 Na2GTP, and 0.1 leupeptin, pH 7.2–7.3, 265–270 mOsm. For the recording of action potentials (APs), whole-cell current-clamp recordings were performed with the internal solution containing (in mM): 125 K-gluconate, 10 KCl, 10 HEPES, 0.5 EGTA, 3 Na2ATP, 0.5 Na2GTP, 12 phosphocreatine, pH 7.25, 280 mOsm. Cells were perfused with ACSF, and membrane potentials were kept at −55 to −65 mV. A series of hyperpolarizing and depolarizing step currents were injected to measure intrinsic properties and to elicit APs. Spontaneous APs were recorded without current injection. For the recording of voltage-dependent sodium and potassium currents, cells (held at −70 mV) were perfused with ACSF, and voltage steps ranging from −90 to + 50 mV were delivered at 10 mV increments. Data analyses were performed with Clampfit (Axon instruments) and Kaleidagraph (Albeck Software).
Real-time quantitative reverse transcription-polymerase chain reaction measurement of gene expression levels
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was performed on total RNA isolated from naivetropic iPSCs grown on gelatin or midbrain neurons differentiated from naivetropic iPSCs. First-strand complementary DNA (cDNA) was synthesized by SuperScript First-Strand Synthesis System (Life Technologies). For each sample, 5 μL cDNA product was used as the template for polymerase chain reaction (PCR) amplification. Reactions were performed in a 25 μL volume with iQ™ SYBR Green Supermix (Bio-Rad) and 200 nM each primers shown in Supplementary Table S1(Supplementary Data are available online at
Statistical analysis
All data were expressed as mean ± standard error of measurement. Unpaired, two-tailed Student's t-tests were performed to evaluate whether the two groups were significantly different from each other. Values of P < 0.05 were considered statistically significant.
TALEN-mediated gene targeting in naivetropic iPSCs
A pair of TALENs were designed according to previously published sequences [29] and assembled with the Golden Gate method (Addgene TALEN Kit #1000000024) [35]. The gene-targeting vector PITX3-2A-eGFP-PGK-Puro [29] was provided by Dr. Rudolf Jaenisch at the Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology. One million naivetropic iPSCs in Human Nucleofector Stem Cell Kit 1 (Lonza) buffer were nucleofected with 5 μg of linearized targeting vector and 4 μg of each TALEN using Nucleofection Program A23. Cells were subsequently plated on DR4 MEF feeders for puromycin selection in naivetropic iPSC medium supplemented with ROCK-inhibitor for the first 24 h. Puromycin selection (0.2 μg/mL) started 48 h later, after nucleofection. Individual colonies were picked up and expanded 7–10 days after puromycin selection. PCR was used to screen for homologous recombinants, which were confirmed by Southern blotting.
Results
Derivation of naivetropic iPSCs from controls and PD patients with parkin mutations
In our previous study, we have generated iPSCs from two normal controls (C001 and C002) and two PD patients with parkin mutations (P001, who has compound heterozygous deletion of exon 3 and exon 5 of parkin, and P002, who has homozygous deletion of exon 3) by reprogramming skin fibroblasts from these subjects with DOX-inducible lentiviruses expressing human Oct4, Sox2, Klf4, c-Myc, and Nanog [10]. As induction of the five transgenes in the presence of 2iL converts primed iPSCs to a naivetropic state [17], we utilized this system to generate patient-specific naivetropic iPSCs. The four lines of primed iPSCs were trypsinized into single cells and replated on MEF feeders in naivetropic culture medium (50% DMEM/F12 and 50% Neurobasal, with N2, B27, 1 mM glutamine, 1% NEAA, 0.1 mM β-mercaptoethanol, penicillin–streptomycin, 5 mg/mL BSA, 2 μg/mL doxycycline, 1 μM PD0325901, 3 μM CHIR99021, and 20 ng/mL human LIF) in an incubator with 5% O2. Some bright and compact cell clumps appeared in the culture 7–12 days later (Fig. 1A). These cell clumps were picked, dissociated into single cells with 0.25% trypsin, and replated on MEF feeders. After 2–3 passages, colonies with mESC-like morphology emerged (Fig. 1B). Further passages of the cells with trypsin generated small, bright, dome-shaped colonies that were very similar to mESCs (Fig. 1C and Supplementary Fig. S1A, J, S, AB). They have been maintained for more than 240 passages (over 2 years) in this culture condition without any significant change in morphology. Furthermore, these naivetropic iPSCs proliferated on gelatin-coated plates in the absence of feeders (Fig. 1D). Immunostaining of the four naivetropic iPSC lines (designated as nC001, nC002, nP001, and nP002) cultured on MEF feeders or gelatin showed that they expressed common pluripotency markers, such as Nanog, Oct4 (Fig. 1E–F and Supplementary Fig. S1B–C, K–L, T–U, AC–AD), and AP (Fig. 1L and Supplementary Fig. S1I, R, AA, AJ), as well as human-specific pluripotency markers such as SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81 (Fig. 1G–J and Supplementary Fig. S1D–G, M–P, V–Y, AE–AH). Despite the similar morphology, they did not express the mouse ESC marker SSEA-1 (Fig. 1K and Supplementary Fig. S1H, Q, Z, AI). In contrast, the AB2.2 mESCs strongly expressed SSEA-1, as well as Nanog, Oct4, and AP, but not SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81 (Fig. 1M–T). Real-time quantitative RT-PCR (qRT-PCR) showed that naivetropic iPSCs expressed endogenous pluripotency genes, including Oct4, Sox2, Klf4, c-Myc, Tert, GDF3, REX-1, DNMT, and TDGF1 (Fig. 1U). Four of the five viral transgenes (Oct4, Sox2, Klf4, and c-Myc) were expressed at variable levels that were high enough to be compared to GAPDH, while Nanog, which serves as the gateway to naive pluripotency [36], was consistently expressed at higher levels (Fig. 1V). RT-PCR of total RNA from the four lines of naivetropic iPSCs confirmed parkin mutations in nP001 and nP002 (Fig. 1W). All four lines of naivetropic iPSCs formed teratomas in SCID mice, demonstrating the pluripotency of these cells (Fig. 1X–Z and Supplementary Fig. S2). G-banding experiments showed that the cells had normal karyotype (Supplementary Fig. S3).
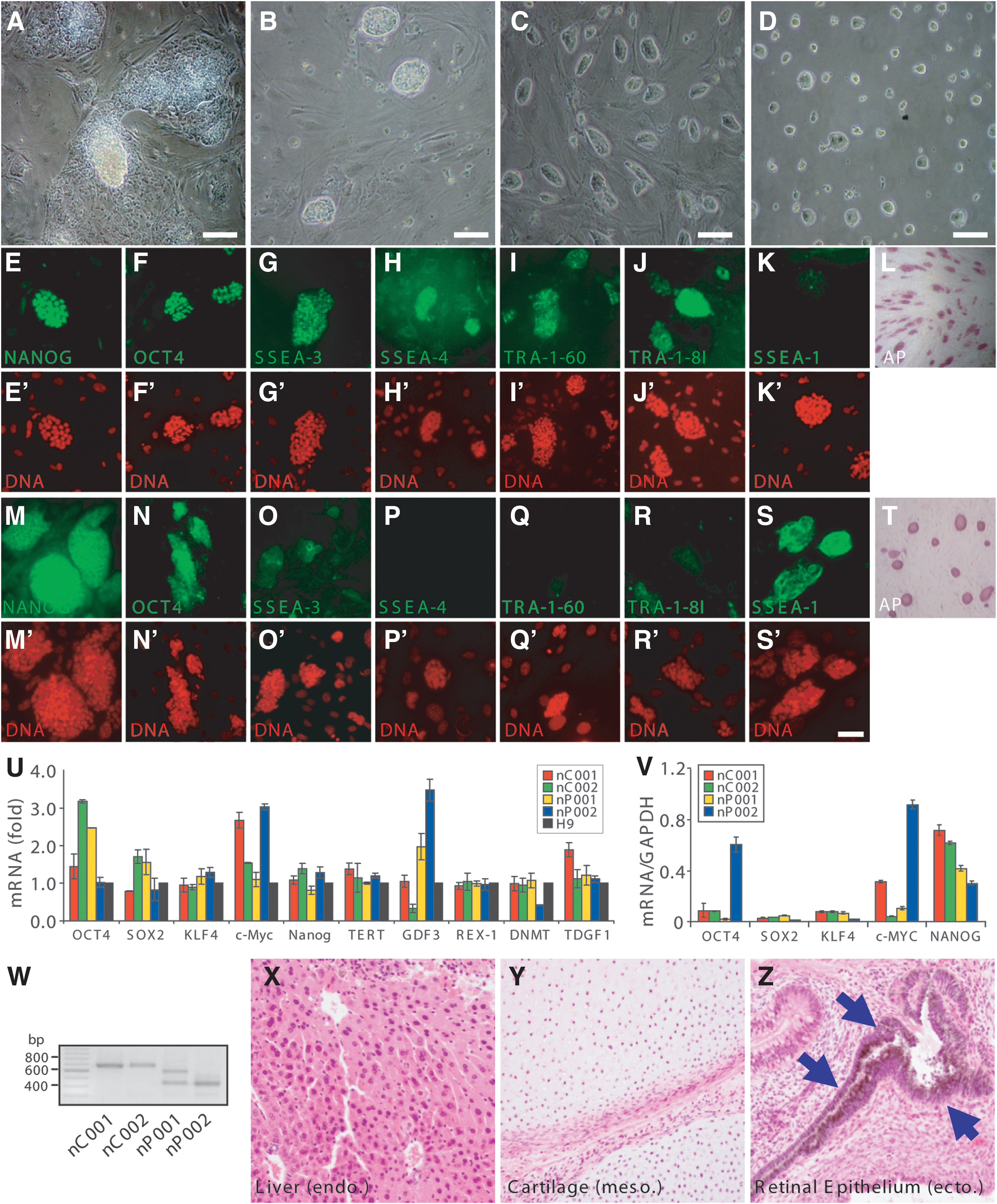
Isolation and characterization of naivetropic induced pluripotent stem cells (iPSCs).
Enriched nuclear localization of TFE3 in naivetropic iPSCs
Exit of naive pluripotency in mouse is gated by the redistribution of TFE3 from the nucleus to the cytoplasm, as the localization of TFE3 shifts from nuclear, in early blastocysts at E3.5, to nuclear and cytosolic in mature blastocysts at E4.5, and then to almost exclusively cytoplasmic in epiblasts at E5.5 [37]. When human primed ESCs are converted to the naive state, TFE3 becomes highly enriched in the nucleus [23]. We costained the parental primed-state iPSCs, the corresponding naivetropic iPSCs, and the primed iPSCs reverted from naivetropic iPSCs (detailed description of these cells later) for TFE3, Oct4, and DAPI. TFE3 was prominently enriched in the nucleus of naivetropic iPSCs (Fig. 2E–H for the P001 series and Supplementary Fig. S4 for the other three series), but showed diffused nuclear and cytosolic localization in the original primed iPSCs (Fig. 2A–D and Supplementary Fig. S4) and the reverted primed iPSCs (Fig. 2I–L and Supplementary Fig. S4). The data corroborate with previous studies on the nuclear enrichment of TFE3 in naive state iPSCs, in comparison to its diffused localization in primed iPSCs.
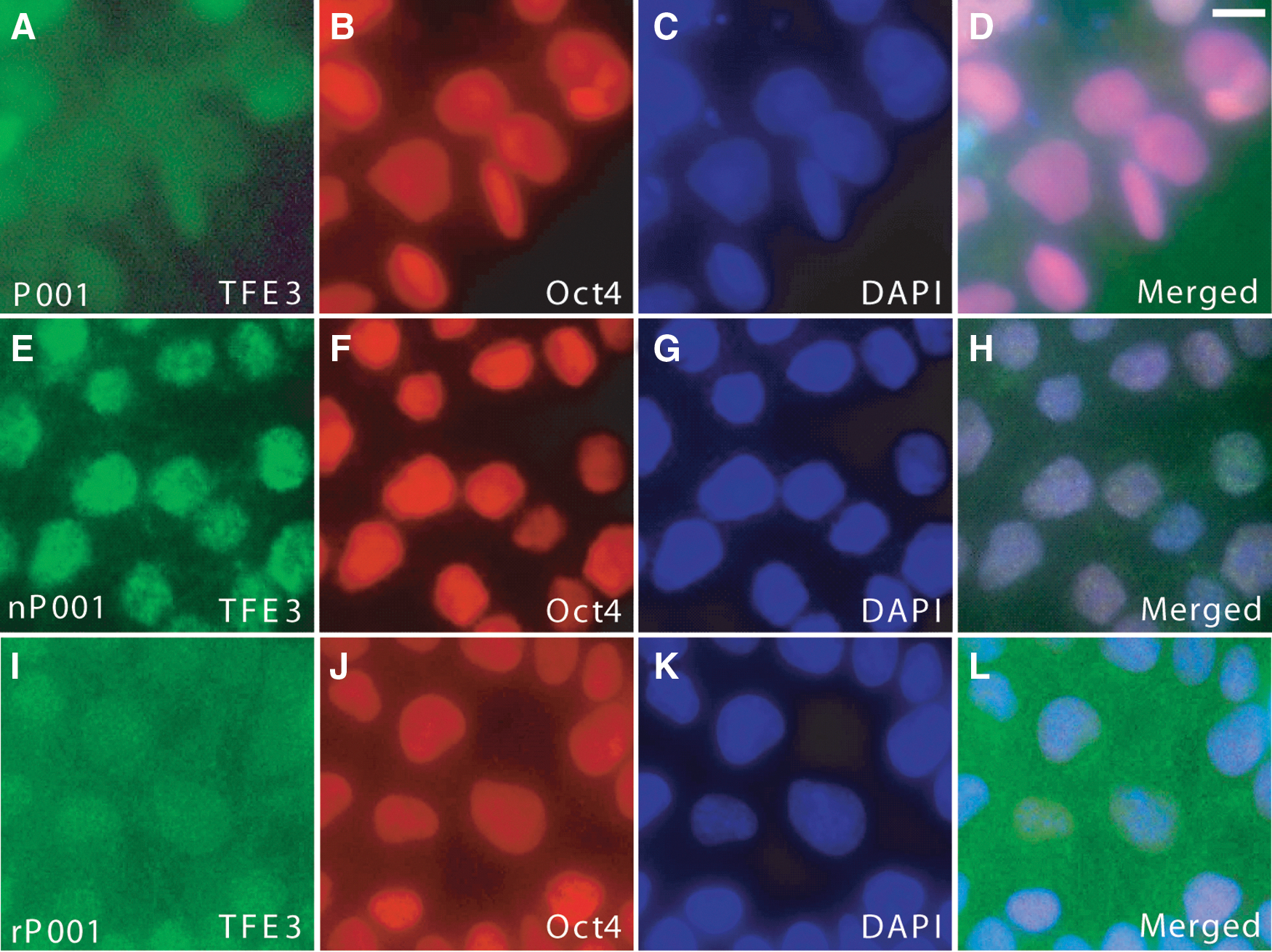
Subcellular localization of TFE3 in primed, naivetropic, or reverted iPSCs.
Patient-specific naivetropic iPSCs exhibit cell doubling time similar to that of mESCs
We noticed that after conversion to the naivetropic state, the iPSCs proliferated much faster. To measure cell doubling time, we plated 10,000 cells from the AB2.2 mouse ESC, naivetropic P002 iPSC (nP002), reverted P002 (rP002, later for details), the parental primed-state P002, or the H9 hESC. For mESC and nP002, trypsinized single cells were plated and the numbers of live PSCs were counted every 3 days. For rP002, P002, and H9, which grew much slower and did not proliferate well after dissociation to single cells, we dissociated the cells in dispase and took a portion of the cell suspension for trypsinization and counting. A volume of cell clamps that contained 10,000 cells were then plated on MEF feeders. The number of live cells was counted every six days using Trypan blue. The growth curve of the five different types of cells clearly showed that there were two groups, mESC and nP002 proliferated at similar rates, which were much faster than the proliferation rates of rP002, P002, and H9 (Fig. 3A). Calculation of cell doubling time showed that it was indeed not significantly different between nP002 (18.6 ± 5.7 h) and mESC (16.3 ± 3.1 h, P > 0.05, n = 3 independent experiments). In contrast, the cell doubling time of primed-state PSCs, including the reverted rP002 (34.2 ± 5.5 h), the parental P002 (36.1 ± 8.1 h), and H9 hESC (35.8 ± 6.2 h), were much longer (P < 0.05, n = 3 independent experiments) (Fig. 3A). We measured cell doubling time of the four lines of patient-specific naivetropic iPSCs and found that they were generally between 18 to 20 h and were not significantly different from each other (P > 0.05, n = 3 independent experiments) (Fig. 3B). Thus, conversion from the primed state to the naivetropic state appears to speed up proliferation of iPSCs significantly.
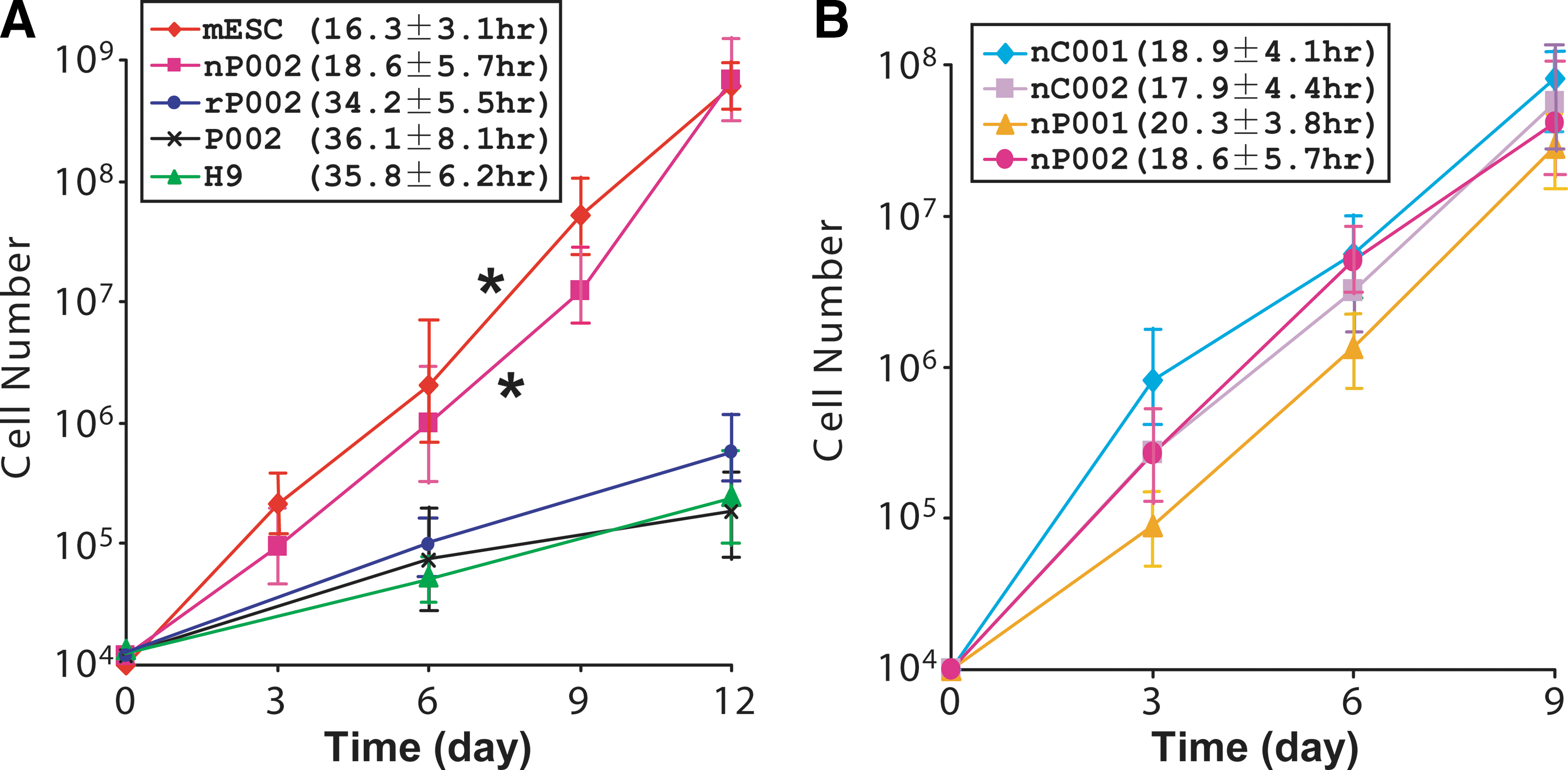
Cell doubling time of naivetropic iPSCs versus other pluripotent stem cells.
Patient-specific naivetropic iPSCs have clonal efficiency similar to that of mESCs
We examined the efficiency of naivetropic iPSCs to form colonies from single cells at 21% or 5% O2 and in the presence or absence of ROCK inhibitor, because hypoxia [38] or ROCK inhibitor [39] increases clonal efficiency of primed iPSCs. Naivetropic iPSC nP002, AB2.2 mESCs, reverted iPSC rP002, or H9 hESCs were trypsinized into single cells and replated on MEF feeders in naivetropic medium or hES medium without or with the ROCK inhibitor Y27632 (10 μM). They were cultured in incubators with 21% or 5% O2 for 3 days for naive PSCs or seven days for primed PSCs. The numbers of PSC colonies were counted by alkaline phosphatase (AP) staining to quantify clonal efficiency, which is defined as the ratio of AP+ colonies to the number of starting cells. The different duration of cell culture was necessary because primed-state PSCs proliferated much slower than naive-state PSCs (Fig. 3). At 21% O2 and in the absence of ROCK inhibitor, clonal efficiency of the naivetropic iPSC nP002 (21.8% ± 4.2%) was much higher than primed-state PSCs such as the reverted rP002 (0.5% ± 0.1%) or H9 hESC (0.2% ± 0.03%). At this condition, clonal efficiency of mESC was 38.7% ± 6.2% (Fig. 4A). The addition of ROCK inhibitor greatly increased clonal efficiency of nP002 to 31.3% ± 3.6%, which was comparable to that of mESC (39.9% ± 5.8%) (Fig. 3B). Both were much higher than the clonal efficiency of primed-state PSCs such as rP002 or H9 hESC (Fig. 4B). Hypoxia (5% O2) culture condition in the absence of ROCK inhibitor significantly increased the clonal efficiency of nP002 (29.8% ± 3.1%, P < 0.05 vs. 21% O2 in Fig. 4A), but did not significantly change the clonal efficiency of mESC (Fig. 4C). The combination of hypoxia and ROCK inhibitor achieved the highest clonal efficiency for nP002 (38.7% ± 6.8%), a level very close to that of mESC (42.3% ± 4.4%) under the same condition (Fig. 4D).
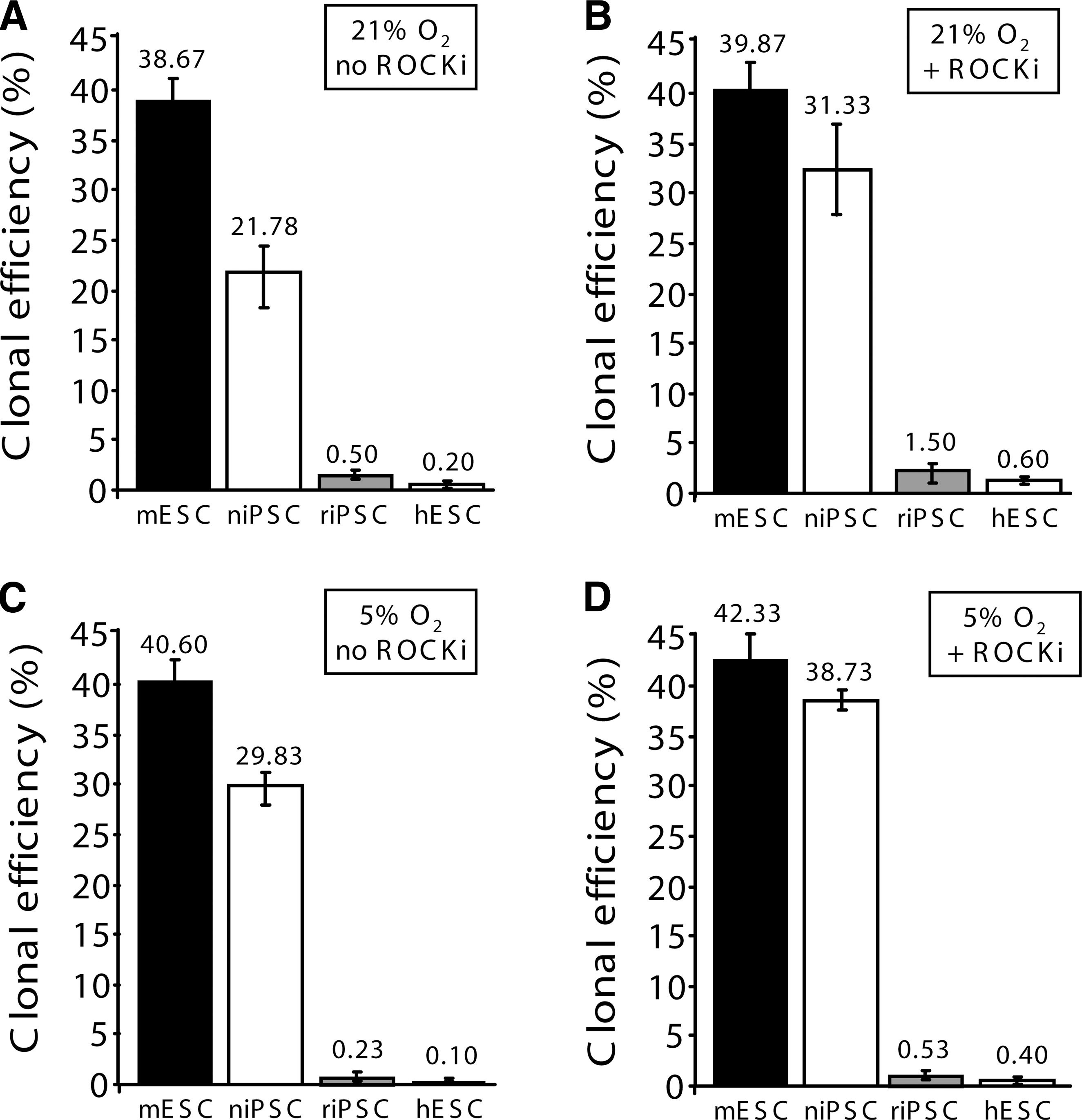
Clonal efficiencies of niPSC, mESCs, riPSC, and hESCs under various conditions.
Patient-specific naivetropic iPSCs can be readily reverted to primed iPSC
Previous studies on transgene-induced naivetropic pluripotency showed that maintenance of the naivetropic state is dependent on the transgenes [17]. We tested whether patient-specific naivetropic iPSCs can be reverted to their original primed state when DOX-mediated transgene expression was withdrawn. Several colonies of nP002 naivetropic iPSCs were manually picked and reseeded on MEF feeders in hES medium with bFGF (4 ng/mL). After 7–10 days, round and flat colonies with typical iPSC morphology emerged (Fig. 5A and Supplementary Fig. S5A, K, U). These iPSCs were designated as reverted iPSCs (riPSC) and the corresponding lines were named rC001, rC002, rP001, and rP002. Immunostaining of these riPSC lines showed that they expressed pluripotency markers, such as Nanog, Oct4, SSEA-3, SSEA-4, TRA-1-81, and TRA-1-60 (Fig. 5B–G for rP002 and Supplementary Fig. S5 for the other three riPSC lines). Spontaneous differentiation assay mediated by EB showed that riPSCs could be differentiated to cells of all three germ layers (Fig. 5H–J for rP002 and Supplementary Fig. S5 for the other three lines). The riPSCs were cultured for over 15 passages in standard hES medium without any significant change in morphology or growth characteristics.
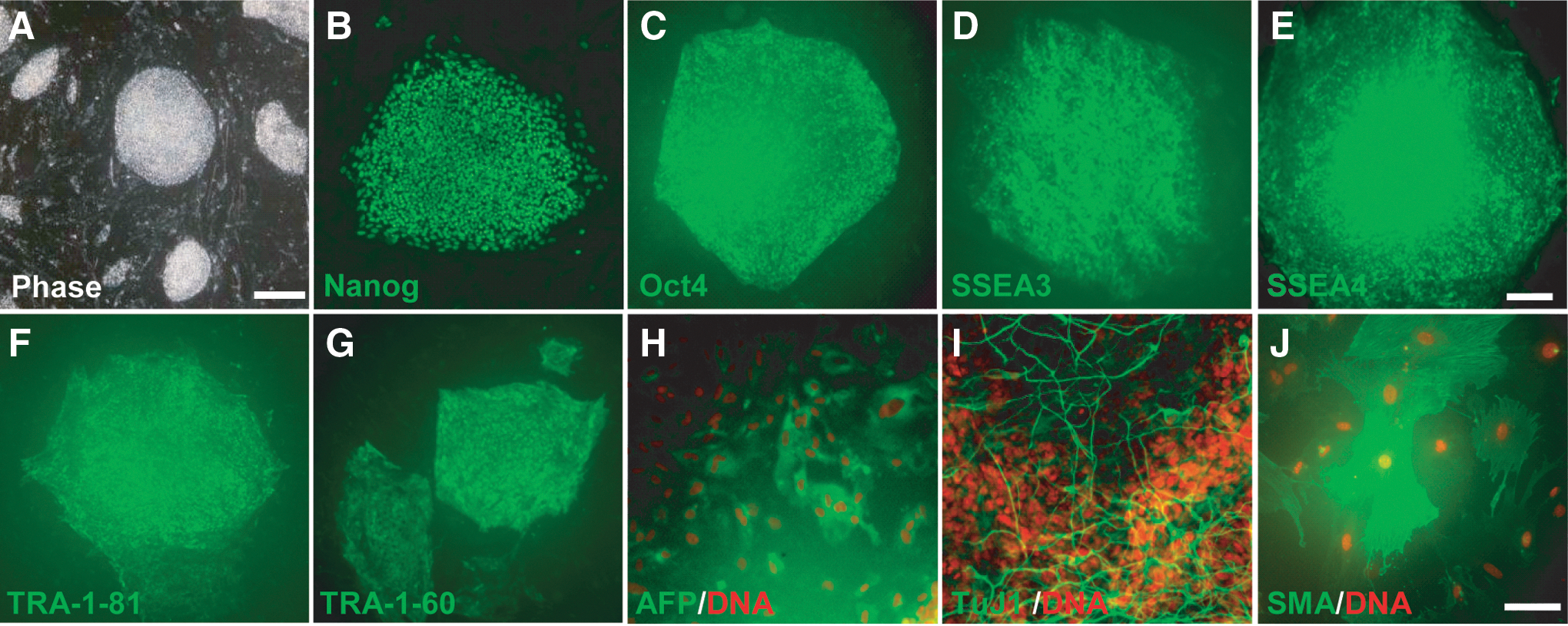
Characterization of iPSCs reverted from naivetropic iPSCs.
Directed differentiation of patient-specific naivetropic iPSCs to midbrain DA neurons
We used a rosette-based directed differentiation protocol [10] to differentiate the four lines of riPSCs to midbrain DA neurons. The riPSC colonies were detached from MEF feeders with dispase and cultured in suspension to form EBs in hESC medium with 10 μM SB431542 for 4 days and then in neural induction medium with 20 ng/mL bFGF for two more days. At day 7, EBs were seeded on laminin-coated plates and treated with FGF8a (20 ng/mL) and SHH (100 ng/mL) until neural tube-like rosettes merged (Fig. 6A). At day 17, rosettes were picked manually and cultured in suspension as neurospheres in neural induction medium with FGF8a and SHH for 7 days (Fig. 6B). At day 24, neurospheres were dissociated to single cells, which were plated on Poly-L-ornithine/Matrigel/laminin-coated plates in neural differentiation medium supplemented with FGF8b, SHH, BDNF, GDNF, and TGF-β3. At day 30–35, neurons with complex morphology were seen (Fig. 6C). Immunostaining of rP002-derived neuronal cultures showed that some of these neurons coexpressed tyrosine hydroxylase (TH) and the midbrain markers engrailed-1 (En-1) (Fig. 6D), AADC (Fig. 6E), VMAT2 (Fig. 6F), Nurr1 (Fig. 6G), and DAT (Fig. 6H). They also coexpressed markers for mature neurons such as MAP2 (Fig. 6I) and synaptic markers such as synaptophysin (Fig. 6J) and NR1 (Fig. 6K). Similar expression of these markers was seen in neuronal cultures derived from rC001, rC002, and rP001 iPSC lines (Supplementary Fig. S6). RT-PCR experiments showed strong and similar expression of marker genes specific for midbrain DA neurons (TH, AADC, VMAT2, DAT, En-1, Pitx3, Nurr1, Lmx1b, and FoxA2) in the four lines of riPSC-derived neurons (Supplementary Fig. S7). Electrophysiological recordings showed that rP002-derived neurons had voltage-gated K+ currents and voltage-gated Na+ currents (Fig. 6L), fire-evoked action potentials (Fig. 5M), spontaneous action potentials (Fig. 6N), and had spontaneous EPSCs (Fig. 6O). Similar electrophysiological profiles were observed in neurons derived from rC001, rC002, and rP001 (Supplementary Fig. S8). These results showed that all four lines of riPSC-derived neuronal cultures contained midbrain DA neurons and the neurons were electrophysiologically and synaptically active.
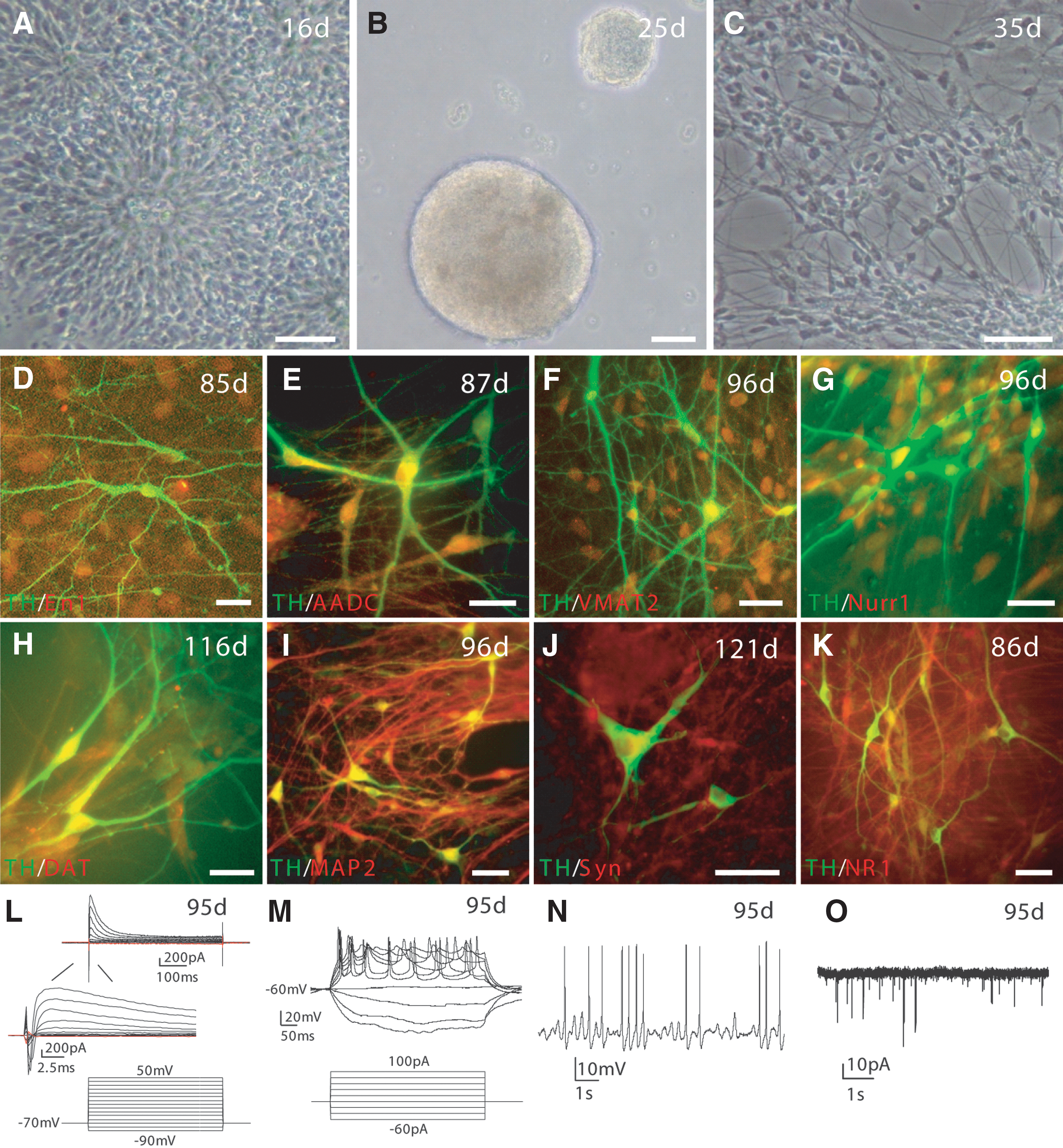
Directed differentiation of rP002 to midbrain dopaminergic (DA) neurons in vitro.
Dopamine-related phenotypes in midbrain DA neurons derived from patient-specific naivetropic iPSCs
To demonstrate that midbrain DA neurons derived from the four lines of naivetropic iPSCs had functional DA transmission, we measured spontaneous and activity-dependent DA release by reverse-phase HPLC coupled with electrochemical detection of dopamine [10]. Midbrain neuronal cultures derived from the four lines of naivetropic iPSCs were incubated at 37°C in HBSS, HBSS with 56 mM KCl, or Ca2+-free HBSS with 56 mM KCl. Spontaneous DA release in HBSS (30 min) was robustly seen in all four lines of DA neurons and was significantly increased in P001 and P002, compared to C001 or C002 (P < 0.05) (Fig. 7A). Membrane depolarization induced by high-concentration KCl (56 mM for 15 min) markedly increased DA release (Fig. 7A). The increase was abolished in Ca2+-free HBSS (Fig. 7A). We calculated Ca2+- and activity-dependent release by the difference between KCl-induced DA release in the presence and absence of Ca2+. It was not significantly different among the four lines of DA neurons (Fig. 7B). To measure specific DA uptake, we incubated the four lines of neurons with 5 μM dopamine in the absence or presence of nomifensine (10 μM), a selective inhibitor of dopamine transporter. The amounts of dopamine in the cells were measured by HPLC. As shown in Figure 7C, specific DA uptake was significantly decreased in DA neurons derived from rP001 and rP002, compared to rC001 or rC002 (P < 0.05, n = 3 independent experiments). The total amounts of endogenous dopamine in the four lines of neurons were not significantly different (Fig. 7D). We also examined the expression levels of MAO-A and MAO-B, which catalyze the oxidative catabolism of dopamine [11]. The mRNA levels of MAO-A (Fig. 7E) and MAO-B (Fig. 7F) were significantly increased in neurons derived from rP001 and rP002, compared to those in rC001 and rC002 (P < 0.01, n = 8). These phenotypes were very similar to what we have observed previously in neurons derived from the parental primed-state iPSCs [10]. Thus, the derivation of naivetropic iPSCs and their subsequent reversion and differentiation do not significantly affect the inherent phenotypes caused by parkin mutations.
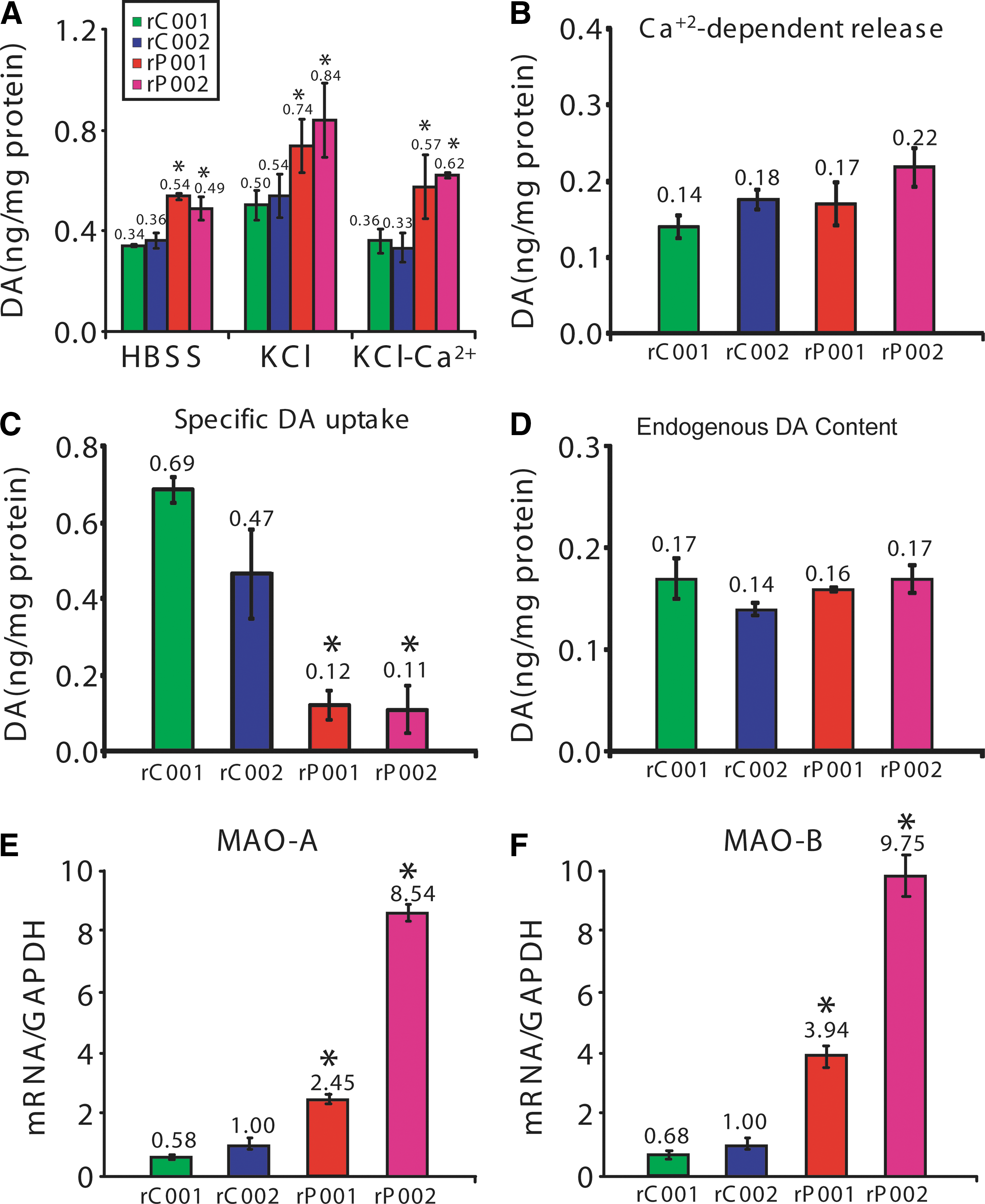
Dopamine-related phenotypes in neurons derived from patient-specific naivetropic iPSCs with parkin mutations.
High-efficiency gene targeting in naivetropic iPSCs using TALEN
The discovery of engineered site-specific nucleases such as TALEN has significantly facilitated gene targeting in human PSCs [29]. However, the very low clonal efficiency and the slow growth rate of primed human PSCs mask the full potential of gene targeting mediated by site-specific nucleases. We tested TALEN-mediated gene targeting in naivetropic iPSCs by using a gene-targeting vector [29] that inserts viral 2A-EGFP sequence in frame with the C-terminus of PITX3 (Fig. 8A), a transcription factor specifically expressed in midbrain DA neurons [40,41]. A pair of TALENs with the same recognition sequences as described previously [29] were generated to introduce a double-stranded break near the stop codon of PITX3 (Fig. 8A). We electroporated 1 × 106 naivetropic iPSCs (nP002 or nC002) with the targeting vector and the TALEN pair. After puromycin selection, clones were manually picked and analyzed first by PCR and then confirmed by Southern blotting (Fig. 8B). Targeting efficiency, which is defined as the percentage of clones with at least one allele targeted, was 64% for nP002 and 67% for nC002 (Fig. 8C). This is much higher than the 21%–23% gene-targeting efficiency achieved in primed human PSCs using the same constructs [29]. In addition, naivetropic iPSCs proliferated much faster, which reduced the duration and cost of gene-targeting experiments.
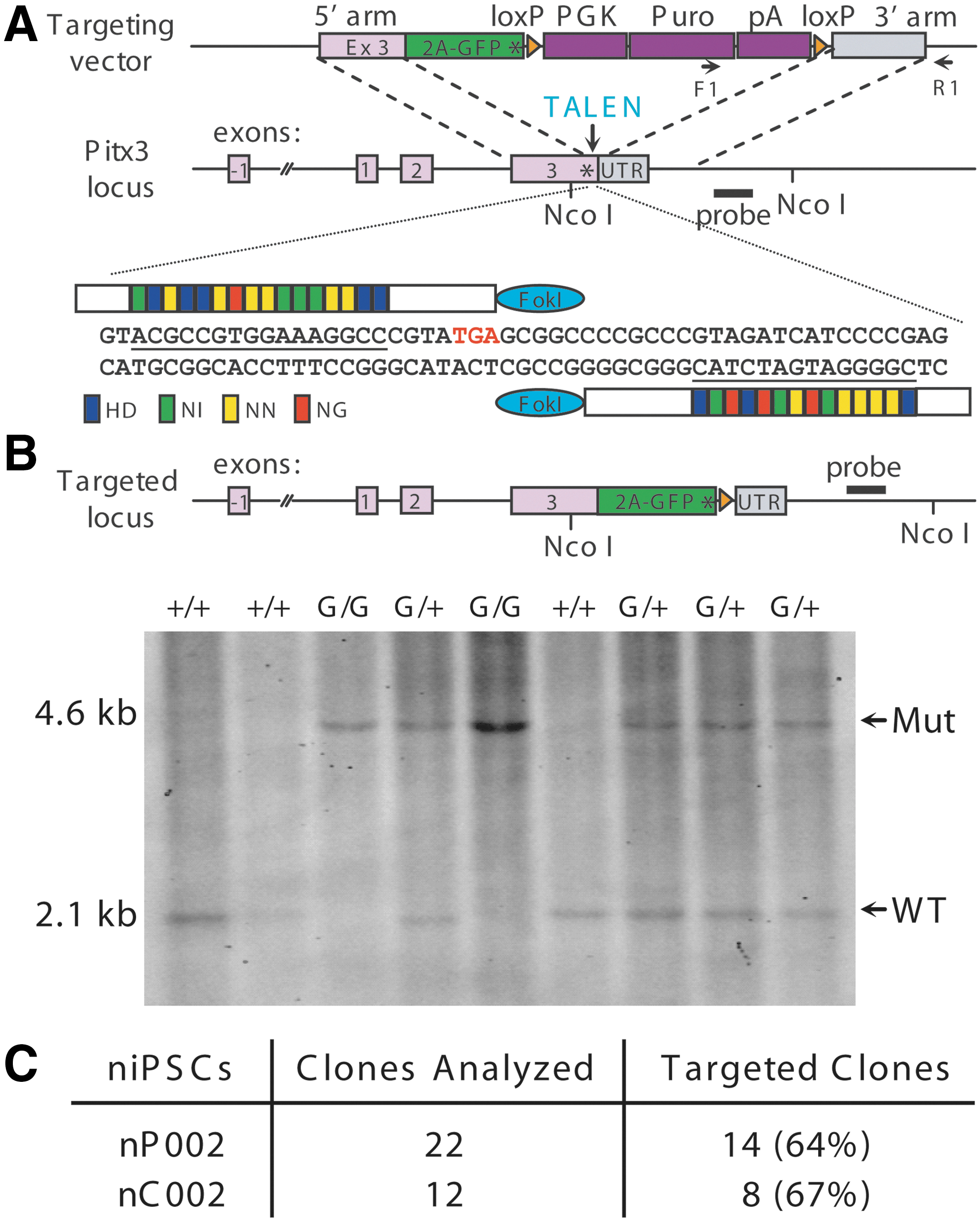
Transcription activator-like effector nuclease (TALEN)-mediated gene targeting of eGFP to the PITX3 locus in naivetropic iPSCs.
Discussion
The discovery of human iPSCs [42,43] has revolutionized mechanistic studies of human diseases, particularly those for which the material of interest is otherwise too invasive to obtain [44]. A case in point is PD, which is caused by the degeneration of nigral DA neurons and many other types of neurons in the brain [1]. The generation of patient-specific iPSCs has enabled investigation of the unique vulnerabilities of midbrain DA neurons from PD patients, particularly those with monogenic mutations in genes such as parkin [10]. Our previous studies have shown that mutations of parkin significantly affect DA neurotransmission and the oxidative catabolism of dopamine by MAOs [10]. To realize the full potential of using patient-specific iPSCs to study PD, we generated naivetropic iPSCs to facilitate precise genetic modifications in these cells.
Indeed, when we targeted eGFP to the PITX3 locus using the same gene-targeting construct and a pair of TALEN that recognizes the same site near the stop codon of PITX3 [29], our targeting efficiency was markedly higher (64%–67% in this study vs. 21%–23% in the previous study) (Fig. 8). The main reason appears to be the significant increase in clonal efficiency when primed iPSCs were converted to the naivetropic state. Even with ROCK inhibitor and hypoxia (5% O2) culture condition, primed iPSCs typically did not survive very well after trypsinization to single cells (Fig. 4D), which is required for gene targeting. Under the same condition, clonal efficiency of naivetropic iPSCs was almost as high as that of mouse ESC (Fig. 4D). When the naivetropic iPSC (niPSC) was reverted to primed iPSC (riPSC), clonal efficiency dropped dramatically back to the level typical for H9 hESC (Fig. 4D). The ability of naivetropic iPSCs to survive after single-cell trypsinization and be able to form clones from a single cell is thus a remarkable feature of naivetropic pluripotency, although the mechanistic detail is unknown at this point. For practical applications such as gene targeting or viral transduction, this feature of naivetropic pluripotency is very useful.
Our results also showed that conversion of primed-state iPSCs to the naivetropic state dramatically sped up cell proliferation. For example, cell doubling time of P002 was shortened from the typical 36.1 h for primed-state iPSCs to 18.6 h, when the same genome was reprogrammed to the naivetropic state in nP002. The cell doubling time was very similar to the 16.3 h for the typical naive mouse ESCs (Fig. 3). When nP002 was reverted to the primed state (rP002), cell doubling time went back to 34.2 h (Fig. 3). This remarkable interconversion is most likely related to the metabolic differences between the two states of pluripotency, which are different epigenetic manifestations of the same genome, as there was no significant genomic change (Supplementary Fig. S3). Naive pluripotency is marked by mitochondrial respiration, which metabolizes glucose much more efficiently than glycolysis—the main metabolic pathway utilized by primed PSCs [45]. The significantly more efficient energy production may allow naive PSCs to proliferate much faster than primed PSCs. The practical corollary is that growth from a genetically modified single cell to a clone would be faster for naive PSCs than for primed PSCs. This would be very useful for gene-targeting experiments.
The main purpose of the present study is to develop a system of patient-specific naivetropic iPSCs for mechanistic study of PD. Using midbrain DA neurons differentiated from naivetropic iPSCs of normal subjects and PD patients with parkin mutations, we found that the phenotypic differences caused by parkin mutations [10] remained the same (Fig. 7). This demonstrates that the epigenetic conversion of primed iPSCs to naivetropic iPSCs and their subsequent reversion and differentiation to midbrain DA neurons did not significantly change the phenotypes that we have identified previously [10]. The robustness of these phenotypes corroborated with the rescue experiments by overexpression of parkin, but not its PD-linked mutant [10]. Increased spontaneous dopamine release (Fig. 7A, B) and reduced dopamine reuptake (Fig. 7C, D) not only disrupt the spatial and temporal precision of DA transmission, but also elevate dopamine concentration in the extracellular domain. Increased transcription of MAO-A and MAO-B (Fig. 7E, F) thus greatly raises the oxidative stress because of the reactive oxygen species produced during the oxidative deamination of dopamine [10].
The biggest advantage of naivetropic iPSCs over their parental lines in the primed state of pluripotency is the high efficiency in which gene targeting can be performed (Fig. 8). This platform will be very useful for dissecting the molecular mechanism of PD using patient-specific midbrain DA neurons. For example, isogenic pairs of naivetropic iPSCs can be generated by repairing parkin mutations in nP001 and nP002 or by introducing parkin mutations to nC001 and nC002. Generation of isogenic pairs of iPSCs that differ only in the absence or presence of parkin mutations will allow us to study the cellular function of parkin in iPSC-derived human midbrain DA neurons without the confounding influence of diverse genetic backgrounds that exist between PD patients and normal subjects. Information gained from the cells would provide definitive answers on how parkin mutations cause the degeneration of human midbrain DA neurons. With the ability to modify the genome at will, we can identify new phenotypes caused by parkin mutations in genetically tagged iPSC-derived neurons, for example, by transplanting them to rodent brains for in vivo study of human midbrain DA neurons. Recent development of transgene-free methods in the derivation of naive human PSCs [23 –27] will make the system even more widely applicable for disease modeling, mechanistic studies, and the development of novel therapies.
Footnotes
Acknowledgments
The authors would like to thank Dr. Rudolf Jaenisch at the Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology for providing the PITX3-2A-eGFP-PGK-Puro gene-targeting construct [![]() ]. The work was supported by the National Key Basic Research Program of China (2011CB504100), NYSTEM contracts C028129, C029556, and C026714, NIH grant NS061856, and Department of Veterans Affairs Merit Award I01BX002452.
]. The work was supported by the National Key Basic Research Program of China (2011CB504100), NYSTEM contracts C028129, C029556, and C026714, NIH grant NS061856, and Department of Veterans Affairs Merit Award I01BX002452.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.