
Abstract
Human induced pluripotent stem cells (iPS cells) resemble embryonic stem cells and can differentiate into cell derivatives of all three germ layers. However, frequently the differentiation efficiency of iPS cells into some lineages is rather poor. Here, we found that fusion of iPS cells with human hematopoietic stem cells (HSCs) enhances iPS cell differentiation. Such iPS hybrids showed a prominent differentiation bias toward hematopoietic lineages but also toward other mesendodermal lineages. Additionally, during differentiation of iPS hybrids, expression of early mesendodermal markers—Brachyury (T), MIX1 Homeobox-Like Protein 1 (MIXL1), and Goosecoid (GSC)—appeared with faster kinetics than in parental iPS cells. Following iPS hybrid differentiation there was a prominent induction of NODAL and inhibition of NODAL signaling blunted mesendodermal differentiation. This indicates that NODAL signaling is critically involved in mesendodermal bias of iPS hybrid differentiation. In summary, we demonstrate that iPS cell fusion with HSCs prominently enhances iPS cell differentiation.
Introduction
I
Cell fusion was used as an approach to study reprogramming of somatic cells toward pluripotency [3,9 –20]. Shortly after fusion, hybrid cells acquire similar characteristics as the parental pluripotent stem cells. Hybrids show the same morphology as the parental pluripotent stem cells and exhibit similar doubling time. They express pluripotency markers, downregulate tissue-specific markers of parental somatic cells, reactivate inactive X chromosome of female somatic cells, and show an undifferentiated epigenetic state [14,21]. They readily differentiate into three germ layers both in vitro and in vivo [15,22]. When injected into diploid blastocysts, hybrids can generate chimeric embryos, with contribution to all the three germ layers and extraembryonic tissues [13,14,22]. Foshay et al. showed that only pluripotent stem cells can induce pluripotency in somatic cells [16]. Factors, which influence reprogramming by cell fusion, include polycomb group proteins [17], activation-induced cytidine deaminase (AID)–dependent demethylation [18], and DNA synthesis [16,19]. However, the differentiation capacity of hybrid cells compared with the parental pluripotent stem cells is less studied, especially in human hybrids. In particular, whether fusion impacts on the differentiation propensity of pluripotent stem cells, such as ES and iPS cells, has remained elusive.
Here, we fused human iPS cells with hematopoietic stem cells (HSCs) from cord blood to generate pluripotent iPS/somatic cell hybrids, in the following referred to as iPS hybrids. We hypothesized that the somatic genome in such hybrids might contribute to their differentiation potential, and thus might serve as an enhancer for iPS cell differentiation.
Materials and Methods
Detailed methods are provided as Supplementary Materials and Methods section (Supplementary Data are available online at
Cell culture and cell fusion
CD34+ HSCs from cord blood were cultured with StemSpan Medium (STEMCELL Technologies) supplemented with 100 ng/mL SCF, 50 ng/mL FLT3 ligand, 20 ng/mL TPO, and 10 ng/mL hyper-IL-6 and infected with Puro-eGFP vector (Supplementary Fig. S1A). iPS cells were obtained from human fibroblasts by transduction with Oct4, Sox2, Klf4, and c-myc in retrovirus or Sendai virus vectors (Supplementary Materials and Methods section). iPS hybrids were produced from iPS cells and HSCs by polyethylene glycol fusion (Fig. 1A) and cells were seeded onto Matrigel-coated culture dishes. Puromycin selection (4 μg/mL; Sigma-Aldrich) was applied 48 h later and hybrid colonies were picked 1 week later and cultured on mouse embryonic fibroblast feeder. The same procedure was used to produce ES hybrids from human H9 ES cells.
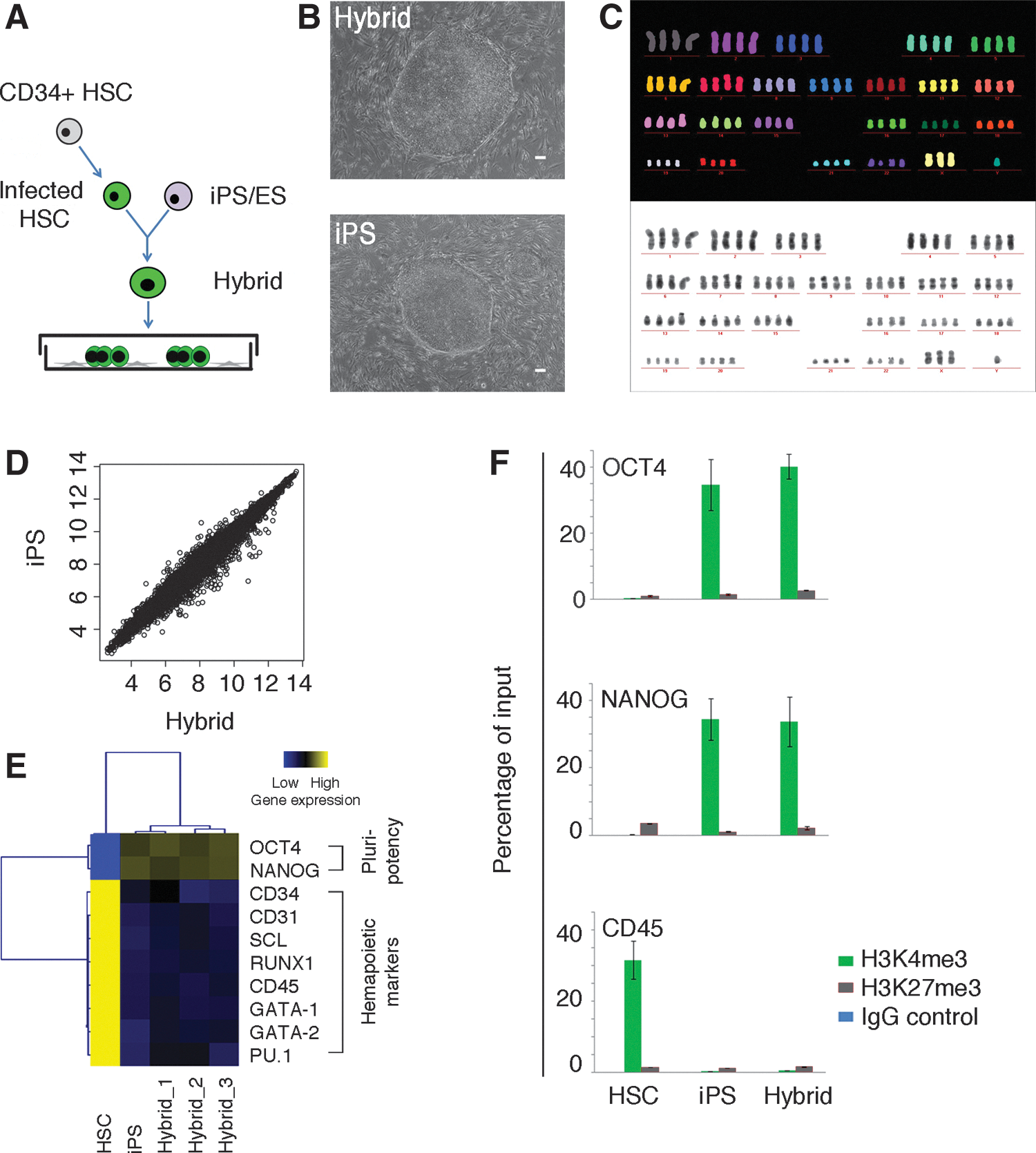
Human induced pluripotent stem (iPS) hybrids are pluripotent.
In vitro differentiation assay
To induce differentiation, undifferentiated cells were dissociated with collagenase IV (20–40 min, 37°C). Cell clusters were harvested and cultured in differentiation medium containing KnockOut DMEM (Invitrogen) and 20% fetal calf serum (FCS; Lonza) in 10-cm ultra-low attachment dishes (Corning Life Sciences). On day 7 of differentiation, embryoid bodies (EBs) were plated on 0.1% gelatin-coated dishes, cultured for another 7–14 days, and then analyzed.
Multicolor-fluorescence in situ hybridization analysis
Cell samples were arrested in metaphase by KaryoMAX Colcemid Solution (0.1 μg/mL, Invitrogen; 2 h, 37°C). Single cells were obtained by trypsin treatment and 75 mM of KCl was added. Samples were centrifuged and the cell pellet was fixed with methanol/acetic acid (3:1, vol:vol). Cells were then subjected to multicolor-fluorescence in situ hybridization (M-FISH) analysis. Briefly, human-chromosome-specific painting probes were combinatorially labeled using seven different fluorochromes and hybridized as previously described [23].
Reverse transcription quantitative polymerase chain reaction, ChIP–quantitative polymerase chain reaction, and microarray analysis
Total RNA was isolated and 1 μg of RNA was used as template for cDNA synthesis. Quantitative polymerase chain reaction (qPCR) was carried out with StepOne Real-Time PCR system (Applied Biosystems). Primer sequences are in Supplementary Table S1. The calculated threshold cycle (Ct) value for each sample was normalized against the corresponding GAPDH value. Expression values were analyzed by using MultiExperiment Viewer (MeV, TM4.7.4;
ChIP-qPCR analysis was performed as described in Dahl and Collas [24]. Briefly, samples were fixed, sonicated, and incubated with polyclonal antibodies to H3K4me3, H3K27me3 (both Diagenode), and normal rabbit serum (Santa Cruz Biotechnology). DNA-protein samples were collected using protein-A Dynabeads (Life Technologies). DNA was prepared and subjected to qPCR analysis as described earlier. Details and primers are listed in Supplementary Materials and Methods section.
For microarray analysis, GeneChip Human Gene 1.0ST Arrays (Affymetrix) were used. Microarray data were preprocessed, normalized by RMA algorithm implemented in Affymetrix Power Tools, and subjected to hierarchical clustering analysis in R. Gene set enrichment analysis (GSEA) was performed by using predefined gene sets of different germ layers [25
–27] and customized R script of GSEA. Data sets were submitted to Gene Expression Omnibus database (
Western blotting
Western blotting was performed according to standard procedures. Membranes were blocked and incubated with anti-p-SMAD2 antibody (Ser465/467, 1:1,000 dilution; Cell Signaling Technology) or anti-ACTIN antibody (Clone AC-74, 1:5,000; Sigma-Aldrich) overnight at 4°C. Membranes were then incubated with peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody (NA934V and NA931, respectively; both 1:5,000; GE Healthcare) for 1 h at RT and bands were detected by chemiluminescence (ECL; GE Healthcare).
Teratoma assay
Undifferentiated cells were harvested with collagenase IV (1 mg/mL, Invitrogen; 40 min, 37°C) and 1–5×106 cells were injected subcutaneously into NOD-SCID mice. Mice were analyzed for teratoma formation 8 weeks later. All experimental procedures involving animal work were approved by local authorities in compliance with the German animal protection law.
Statistical analysis
All data were obtained from at least three independent experiments. Statistical analysis of the data was performed with unpaired Student's t-test. P<0.05 was considered statistically significant. Data are shown as mean±SD.
Results
iPS hybrids show mesendodermal differentiation bias
Hybrids from iPS cells and HSCs showed iPS cell morphology and were tetraploid (Fig. 1B, C and Supplementary Fig. S1B). iPS hybrids had similar cell proliferation rates as parental iPS cells (Supplementary Fig. S1C). They expressed the pluripotency markers OCT4, stage-specific embryonic antigen-4 (SSEA4), and tumor rejection antigens 1-60 (TRA-1-60) similar to parental iPS cells (Supplementary Fig. S1D). Further, iPS hybrids were very similar to parental iPS cells in global gene expression (Fig. 1D), including upregulation of the pluripotency markers OCT4 and NANOG and downregulation of the hematopoietic markers CD34, CD45, and RUNX1 (Fig. 1E). Accordingly, OCT4 and NANOG promoter regions of iPS hybrids showed the transcriptionally active mark H3K4me3, similar to parental iPS cells, as determined by chromatin immunoprecipitation (ChIP) analysis (Fig. 1F). The hematopoietic marker CD45 showed the H3K4me3 mark in HSCs, which was lost in iPS hybrids. To characterize the differentiation capacity of iPS hybrids, we performed EB and teratoma assays. Tissue types of all three germ layers were found in EBs and teratomas of hybrids, indicating that iPS hybrids had three-germ-layer differentiation capacity both in vitro and in vivo (Supplementary Fig. S1E, F). Taken together, we demonstrate that iPS hybrids were pluripotent.
Interestingly, EBs of iPS hybrids showed prominent cystic structures (day 7), which were not observed or only found at low frequency in parental iPS cells (Fig. 2A). Mesodermal markers, such as CD31, CD34, HCN4, and α-myosin heavy chain (α-MHC), and endodermal markers alpha-fetoprotein (AFP) and albumin (ALB) were more abundantly expressed in iPS hybrids compared with parental iPS cells (days 7 and 14; Fig. 2B and Supplementary Fig. S2A). Ectodermal markers, such as SRY-box containing gene 1 (SOX1) and microtubule-associated protein 2 (MAP2), were three- to fourfold less expressed in differentiating iPS hybrids than in parental iPS cells (Fig. 2B and Supplementary Fig. S2A). Similar results were obtained by immunohistochemical staining of EB sections and teratomas (Supplementary Fig. S2B, C). Genome-wide gene expression profiling during differentiation demonstrated that iPS hybrids clustered distinctly from iPS cells (Fig. 2C). Specifically, GSEA on key transcription factors of three-germ-layer differentiation showed a mesendodermal differentiation bias of iPS hybrids already at the initial stages of differentiation (days 2 and 4; Fig. 2D). Surprisingly, when we subjected iPS hybrids to neural-specific differentiation, they maintained the bias toward more mesendodermal progenies, while parental iPS cells readily differentiated into neural lineages (Fig. 2E). In addition, we found the mesendodermal differentiation bias in both early and late-passage iPS hybrids (P10 and P35, respectively; Fig. 2F).
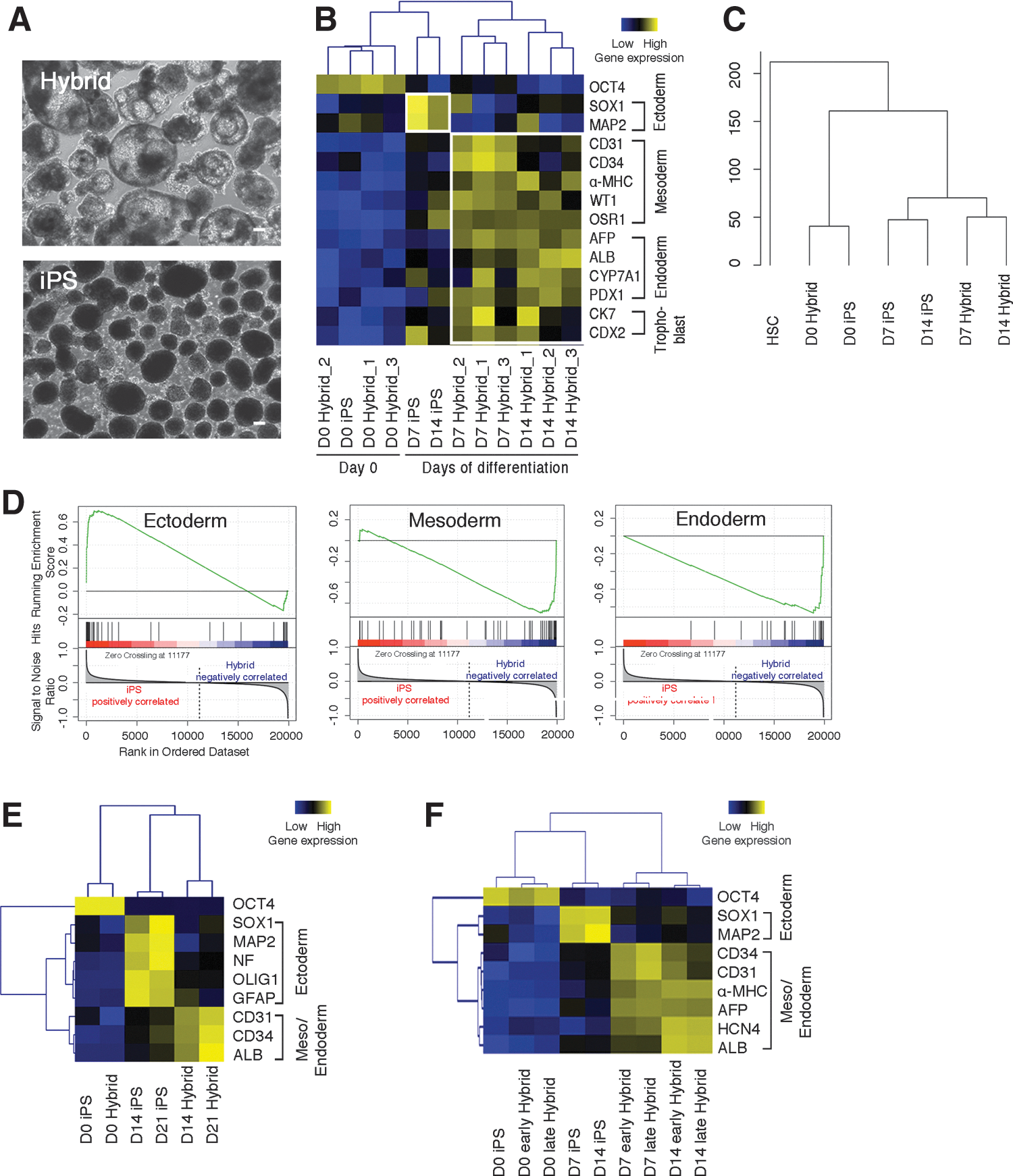
iPS hybrids show differentiation bias toward mesendoderm.
Progenies of iPS hybrids are fully functional
We then proceeded to analyze the differentiated cell types of iPS hybrids. iPS hybrids gave rise to erythroid and myeloid colonies under hematopoietic differentiation conditions, while colony formation of parental iPS cells was rather poor (Supplementary Fig. S3A–C and data not shown). Additionally, more CD34+CD43+ and CD34+CD45+ hematopoietic progenitors were obtained from iPS hybrids than from iPS cell control (Supplementary Fig. S3D, day 14). Hematopoietic progenitor cells were then differentiated into dendritic cells (DCs) with FLT3 ligand (Supplementary Fig. S3A). iPS hybrids generated more CD11c+ DCs than parental iPS cells (52% and 9%, respectively; Supplementary Fig. S3E, day 21). The enhanced hematopoietic differentiation potential of iPS hybrids is very much in line with gene expression data of hematopoietic progenitors (CD34, CD43, and CD45) and DCs (PU.1, FLT3) by reverse transcription-qPCR (Supplementary Fig. S3F).
Further, endothelial cells derived from iPS hybrids showed the capacity of low-density lipoprotein uptake and tube formation (Supplementary Fig. S4A, B). Beating areas, indicating functional cardiomyocyte derivation, were found at a higher frequency in differentiating iPS hybrids than in parental iPS cells (day 14; Supplementary Fig. S4C and Supplementary Video). Hepatocytes derived from iPS hybrids showed the ability of urea secretion and glycogen synthesis (Supplementary Fig. S4D, E). Taking together, iPS hybrids readily differentiated into a variety of functional cells and showed a prominent differentiation bias toward mesendoderm.
iPS hybrids generated from different cord blood samples and different iPS cell lines consistently showed similar mesendodermal differentiation bias (Supplementary Fig. S5A–C). As a further control, ES hybrids were generated from human H9 ES cells and HSCs (Supplementary Fig. S6A–C). In EB assays, such ES hybrids showed impaired neural differentiation compared with parental ES cells (Supplementary Fig. S6D), as was observed for iPS hybrids. ES hybrids also showed prominent mesendodermal differentiation propensity under neural differentiation condition (Supplementary Fig. S6E). Collectively, mesendodermal differentiation propensity of hybrids is independent of HSC preparations and pluripotent cell lines.
Differentiating iPS hybrids exhibit prominent and accelerated NODAL signaling
To analyze the underlying mechanism of this mesendodermal differentiation propensity, we measured the expression kinetics of T, MIXL1, and GSC, which are early transcription factors of primitive streak for mesendoderm commitment [28,29]. iPS hybrids expressed all these markers at higher levels than parental iPS cells with a prominent peak at day 2 (Fig. 3A). Additionally, these expression data correlated well with an enrichment of the active mark H3K4me3 at the T, MIXL1, and GSC promoters at day 2 (Fig. 3B). In parental iPS cells, there was no such enrichment at day 2 (data not shown). NODAL, an important gene for mesendodermal differentiation [29 –31], showed an accelerated kinetics and a more prominent expression in differentiating iPS hybrids compared with iPS cell control (Fig. 3C). This was accompanied with an enrichment of the active mark H3K4me3 at the NODAL promoter at day 2 (Fig. 3D). The NODAL downstream signaling protein phosphorylated-SMAD2 (p-SMAD2) also occurred with accelerated kinetics at day 2 in iPS hybrids compared with parental iPS cells (Fig. 3E). Collectively, we show that ACTIVIN/NODAL signaling, which is a key component in mesendodermal commitment, is highly activated at an early stage of iPS hybrid differentiation.
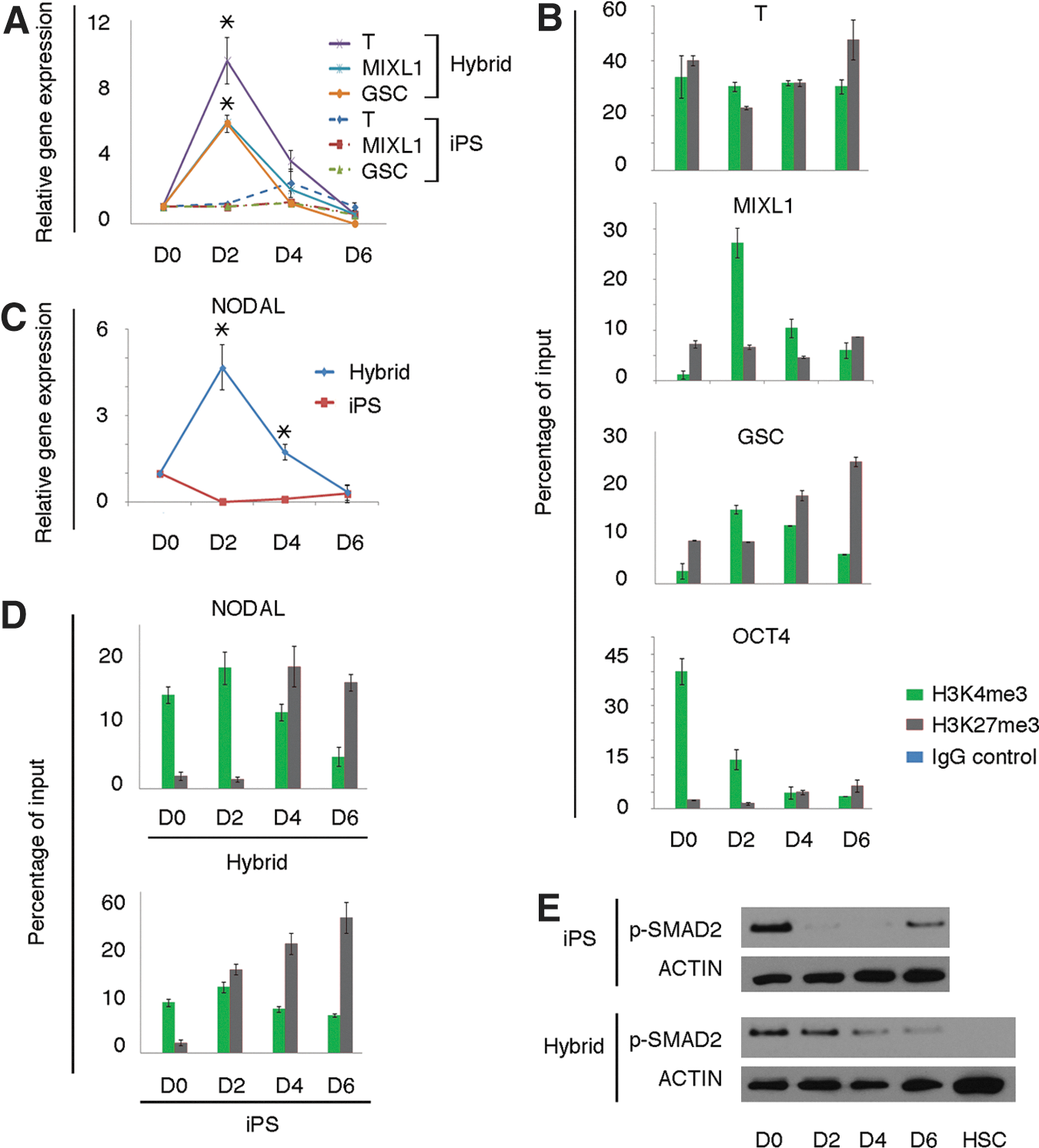
Upregulation of early mesendoderm markers in iPS hybrids.
To test the impact of NODAL on the mesendodermal differentiation propensity in iPS hybrids, we applied SB431542 (SB) to block ACTIVIN/NODAL/SMAD signaling. SB efficiently inhibited p-SMAD2 and the occurrence of cystic EBs (Fig. 4A–C). Primitive streak markers and lineage markers from mesendoderm were downregulated, while ectodermal markers were upregulated, indicating that the propensity of mesendodermal differentiation in iPS hybrids shifted toward ectoderm (Fig. 4D, E). Essentially the same result was obtained when applying SB in EB assays under serum-free conditions (Fig. 4F). Further, enhanced expression of primitive steak markers and NODAL signaling was also observed in other iPS hybrids (iPS_2 hybrids, Supplementary Fig. S7A–C). In summary, these findings suggest that the active ACTIVIN/NODAL/SMAD signaling is a crucial component of the mesendodermal propensity in iPS hybrid differentiation.
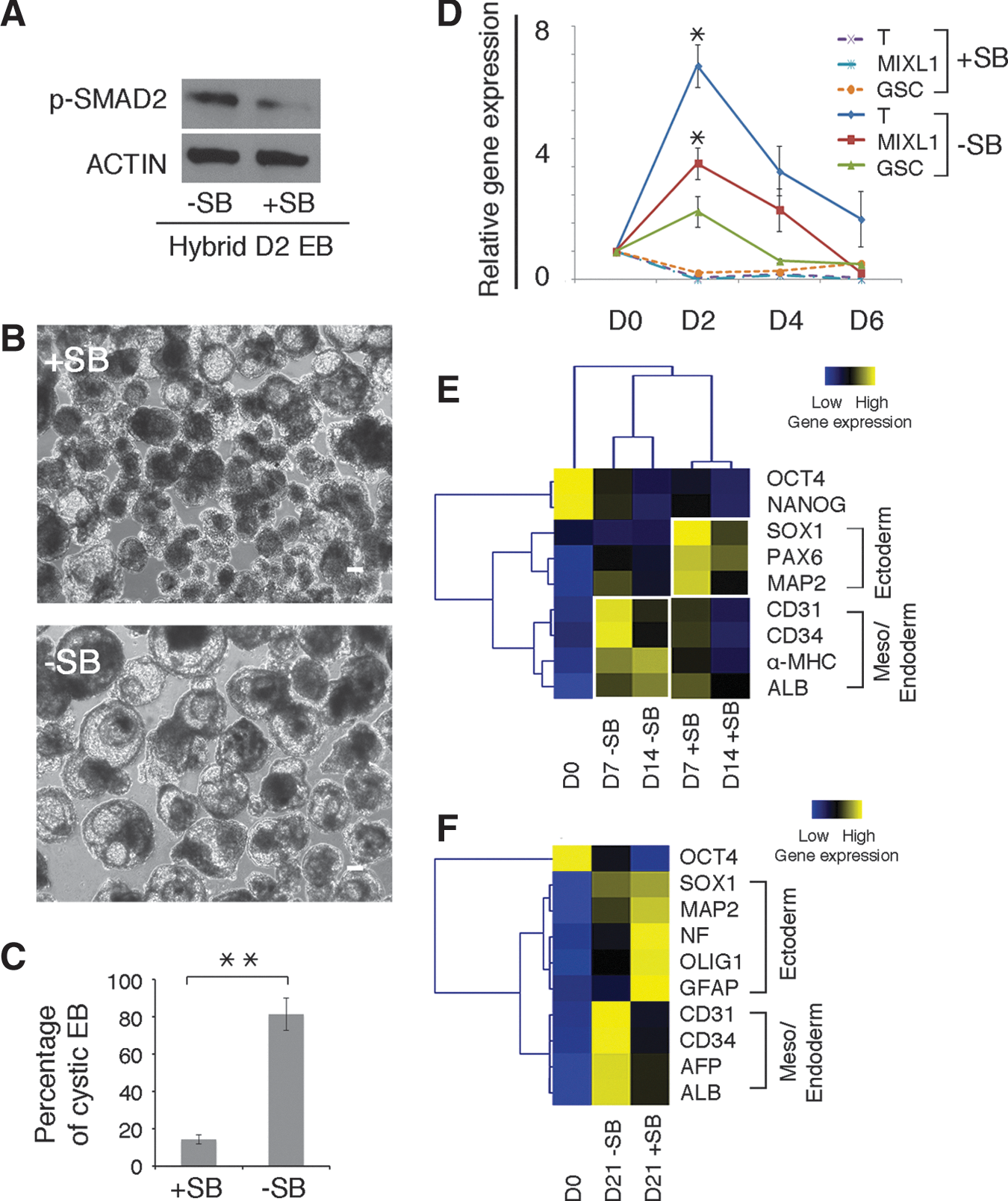
Mesendodermal differentiation bias of iPS hybrids is blocked by ACTIVIN/NODAL inhibitor SB431542 (SB).
Discussion
In this study, we demonstrate that iPS hybrids with HSCs show mesendodermal differentiation bias. Such iPS hybrids harbor enhanced differentiation capacity toward the hematopoietic lineage and also toward other mesendodermal lineages (endothelium, cardiomyocytes, and hepatocytes). Similarly, ES hybrids with HSCs also showed a mesendodermal differentiation bias. This is interesting, since neuroectoderm differentiation is considered to be the “default” pathway of in vitro differentiation of human pluripotent stem cells [32]. Thus, cell fusion of pluripotent stem cells with somatic cells provides a means to direct their differentiation propensity.
Mesoderm and endoderm originate from primitive streak and this is regulated by NODAL, WNT3, and BMP4 signaling [29,33]. NODAL is the major factor in primitive streak formation and mesendoderm development [29,30,33]. Here, we found that NODAL signaling is the main trigger of mesendodermal differentiation bias of iPS hybrids. Mesendoderm development is guided by the combined effect of several signaling pathways, including WNT3 and BMP4. Accordingly, we observed also an upregulation of WNT3 and BMP4 during differentiation in some iPS hybrid clones, which was associated with prominent mesodermal differentiation propensity (data not shown). This suggests that signaling pathways other than NODAL can participate in enhancing mesendodermal differentiation of iPS hybrids.
One interesting question is that whether the enhanced NODAL signaling in iPS hybrids is the result of “somatic memory” originating from the somatic fusion partner? Somatic memory as remnants of the somatic cell has been described in iPS cells [34,35]. Somatic memory causes a differentiation bias toward lineages of the somatic cells used for reprogramming. In addition, with continuous passaging the somatic memory of iPS cells is erased [34,36].
The differentiation bias of iPS hybrids appears to be different from the somatic memory in iPS cells. First, iPS hybrids showed a differentiation bias toward a broad range of cell types, comprising hematopoietic cells and also other mesendodermal cell types. Second, in iPS hybrids, the mesendodermal differentiation bias persisted for at least 34–40 passages, whereas the somatic memory of iPS cells vanishes with higher passage numbers (passage >12) [36]. Finally, HSCs do not express NODAL, which is the main trigger of the mesendodermal differentiation bias of iPS hybrids. As a result, the mesendodermal differentiation bias of iPS hybrids cannot simply be explained by remnants of the HSC fusion partner. So far, we cannot exclude the possibility that other unknown somatic memory factors and/or mechanisms contribute to the differentiation bias of iPS hybrids [37].
There is also the possibility that the differentiation bias observed here is a distinct trait of the 4n status of the hybrids. Previous studies performed in vitro differentiation assay of hybrids to show three-germ-layer differentiation potential, but did not determine the differentiation capacity toward individual germ layers [9,14]. Experiments of mouse chimeras with cell aggregates of hybrids and normal diploid cells revealed integration of hybrid cells into a limited number of organs of the embryos (liver, heart, kidney, and gut) [21,38]. A competitive growth pressure between hybrid cells and diploid cells might account for this observation [39]. Alternatively, this could also be the result of the differentiation bias of the hybrid cells as observed here.
Taken together, our results extend previous studies [3,9 –19], where somatic cells were fused with ES cells to study reprogramming and induction of pluripotency. Here we show that fusion with somatic cells impacts on the differentiation propensity of pluripotent stem cells. In addition, our findings provide a strategy for enhancing iPS cell differentiation toward those cell types, which are notoriously difficult to obtain.
Footnotes
Acknowledgments
The authors would like to thank C. Becker, P. Wanek, H. Chauvistré, and H. Holtgreve-Grez for technical assistances. We also want to thank A. Offergeld and R. Sous for expert secretary assistance. This work was supported by the StemCellFactory consortium, cofunded by the European Union (European Regional Development Fund–Investing in your future) and German Federal State of North Rhine-Westphalia (M.Z., O.B., and W.W.), the Stem Cell Network NRW (W.W.), National Natural Science Foundation of China grants 30911130363 (R.C.Z.), German Federal Ministry of Education and Research (BMBF) grant no. 1315799 (O.B.), donation U. Lehman and funds from German Research Foundation (DFG) grants ZE432/5-2 and ZE432/6-1 (M.Z.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.