
Abstract
Mesenchymal stromal cells (MSCs) are promising tools for therapeutic revascularization of ischemic tissues and for support of vessel formation in engineered tissue constructs. Recently, we could show that avascular-derived MSCs from placental amnion release soluble factors that exhibit survival-enhancing effects on endothelial cells (ECs). We hypothesize that MSCs derived from placental blood vessels might have even more potent angiogenic effects. Therefore, we isolated and characterized MSCs from placental chorionic blood vessels (bv-MSCs) and tested their angiogenic potential in comparison to amnion-derived avascular MSCs (av-MSCs). bv-MSCs express a very similar surface marker profile compared with av-MSCs and could be differentiated toward the adipogenic and osteogenic lineages. bv-MSCs exert immunosuppressive properties on peripheral blood mononuclear cells, suggesting that they are suitable for cell transplantation settings. Conditioned medium (Cdm) from av-MSCs and bv-MSCs significantly enhanced EC viability, whereas only Cdm from bv-MSCs significantly increased EC migration and network formation (Matrigel assay). Angiogenesis array analysis of av- and bv-MSC-Cdm revealed a similar secretion pattern of angiogenic factors, including angiogenin, interleukins-6 and -8, and tissue inhibitors of matrix metalloproteinase-1 and 2. Enzyme-linked immunosorbent assay analysis showed that, in contrast to av-MSCs, bv-MSCs secreted vascular endothelial growth factor. In direct coculture with bv-MSCs, ECs showed a significantly increased formation of vessel-like structures compared with av-MSCs. With regard to therapeutic treatment, bv-MSCs and particularly their Cdm might be valuable to stimulate angiogenesis especially in ischemic tissues. av-MSCs and their Cdm could be beneficial in conditions when it is required to promote the survival and stabilization of blood vessels without the risk of unmeant angiogenesis.
Introduction
M
MSCs are commonly isolated from bone marrow or other adult tissues, such as adipose tissue. This complicates their use due to invasive isolation methods and impaired proliferation and differentiation capacities, which possibly depend on the age and disease stage of the donors [9,10]. MSCs isolated from postnatal tissues, such as placenta (including fetal membranes), umbilical cord, and cord blood, are therefore appealing alternative cell types.
The amnion forms the inner avascular layer of the fetal membranes and is an especially promising source of cells for therapeutic use. Its first clinical application was reported more than 100 years ago as a surgical material in skin transplantation [11]. Since then, it has been applied in various medical conditions, including chemical burns, skin ulcers, and ophthalmology. Its beneficial effects are assigned to its anti-inflammatory, immunomodulatory, and scar-formation-reducing properties [12]. Even though the exact mechanisms are not known yet, secreted factors are suggested to play an important role [13].
We could recently show that amnion-derived MSCs release soluble factors that exhibit beneficial, survival-enhancing effects on endothelial cells (ECs), in spite of the fact that the amnion is an avascular tissue [14]. We hypothesize in the current study that MSCs from a perivascular origin might have even more potent angiogenic effects. Therefore, we isolated and characterized MSCs from placental chorionic blood vessels (bv-MSCs) and tested their angiogenic potential in comparison to amnion-derived avascular MSCs (av-MSCs). We collected conditioned medium (Cdm) from both cell types and investigated its effect on EC viability, network formation, and migration. As low-oxygen concentrations are known to induce angiogenesis [15] and have a proangiogenic effect on MSCs [16], we collected Cdm from cultures at 2% in addition to 21% oxygen. Further, we identified possible angiogenic factors in Cdm using an angiogenesis array and enzyme-linked immunosorbent assay (ELISA) and also investigated direct effects of MSCs on ECs in coculture settings.
Materials and Methods
Sample collection
Human term placentas of normal pregnancies (range 38–42 weeks) were obtained from women after spontaneous delivery or cesarean section at the Department of Gynecology and Obstetrics at the University Hospital Graz. The study received local ethical approval (No. 21-079 ex 09/10) and all women gave written informed consent. Placental tissues were immediately transported to the laboratory for isolation of MSCs from avascular tissue (av-MSCs, n=10) and blood vessels of the chorionic plate (bv-MSCs, n=3). ECs were isolated from placental arterial blood vessels (n=3).
Isolation and culture of av-MSCs
av-MSCs were isolated from the placental amnion as described earlier [14,17]. Amnion and chorion were manually separated and washed with 0.9% saline (Fresenius Kabi) supplemented with 150 IU/mL penicillin, 150 μg/mL streptomycin (both from PAA Laboratories), and 0.4 μg/mL amphotericin B (Gibco, Invitrogen). The amnion was cut into small pieces and incubated with 2.5 U/mL dispase (BD Biosciences) for 9 min at 37°C. The pieces were transferred into Dulbecco's modified Eagle's medium (DMEM) low glucose (Gibco, Invitrogen) supplemented with 15% fetal bovine serum (FBS) Gold (Gibco, Invitrogen), 100 IU/mL penicillin, and 100 μg/mL streptomycin for 10 min. Subsequently, 1.0 mg/mL of collagenase A and 0.01 mg/mL of DNase were added for 2 h (both from Roche). After centrifugation for 3 min at 150 g, the supernatant was poured through a cell strainer (100-μm mesh size; BD Biosciences) and centrifuged for 10 min at 300 g. The pellet was washed with phosphate-buffered saline (PBS; Gibco, Invitrogen) and resuspended in 10 mL of endothelial growth medium-2 (EGM-2; Lonza). EGM-2 contains 2% FBS, epidermal growth factor (EGF), hydrocortisone, vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), insulin-like growth factor 1, ascorbic acid, and heparin. After a further centrifugation step (10 min at 300 g) the cell pellet was resuspended in 10 mL EGM-2 and cells were cultured on 1% gelatin (PAN-Biotech GmbH)–coated flasks. Medium was changed every 2–3 days.
Isolation and culture of bv-MSCs
To isolate bv-MSCs, chorionic blood vessels from the fetal surface of the chorionic plate of term placentas were extracted and short pieces of ∼8 mm in length were resected and transferred to culture plates coated with 1% gelatin. Each vessel explant was carefully adhered to the bottom and cultured separately in one well (of a six-well plate) with 1 mL of EGM-2. One milliliter of medium per well was added the next day and media were then changed every 2–3 days. After 4–7 days, the tissue explants were removed and the outgrown cells were further cultured in EGM-2. The cell identity was confirmed by immunocytochemistry. Only cell cultures with positive staining for the fibroblast marker CD90 (CD90, clone ASO2, 0.1 μg/mL, mouse IgG1; Dianova) and absence of markers against smooth muscle actin (smA, 1A4, 0.2 μg/mL, mouse IgG2a; Dako), desmin (D33, 0.4 μg/mL, mouse IgG1; Dako), and the endothelial marker von Willebrand factor (vWF; immunoglobulin fraction, rabbit anti-human, 0.7 μg/mL; Dako) were used for further experiments or frozen until further use.
Isolation and culture of placental ECs
ECs from normal term human placentas were isolated as published earlier [18]. Briefly, after removal of the amnion, arterial chorionic blood vessels at the fetal surface of the chorionic plate were resected. Vessels were washed with Hank's balanced salt solution (HBSS; Gibco, Invitrogen) to remove residual blood. ECs were isolated by perfusion of vessels with HBSS containing 0.1 U/mL collagenase and 0.8 U/mL dispase (both from Roche), supplemented with 300 IU/mL penicillin and 300 μg/mL streptomycin, prewarmed to 37°C. The perfusion time was limited to 7 min to avoid contamination with non-ECs. The cell suspension was centrifuged (200 g for 5 min) and the pellet was resuspended with EGM-MV medium (Lonza). The cells were plated on culture plates precoated with 1% gelatin and cultured in EGM-MV medium. The endothelial identity was confirmed by positive staining for the classical endothelial marker vWF and absence of markers against fibroblasts (CD90) and smooth muscle cells (smA and desmin). For all experiments ECs were used in passage 3.
Flow cytometry analysis
av- and bv-MSCs were harvested and immediately processed for flow cytometric analysis as previously described [19]. Briefly, cells were washed with PBS twice, blocked with sheep serum (10% v/v, 30 min, 4°C), and stained with directly fluorochrome-labeled mouse anti-human monoclonal antibodies [HLA-DR, CD13, CD14, CD19, CD31, CD34, CD45, CD49a, CD63, CD73, CD166, MSCA1, HLA-DR, alkaline phosphatase (AP; all from BD), CD90 (Beckman Coulter), CD105 (Caltag Laboratories, Burlingame, CA), HLA-ABC (Harlan Sera-Lab), and CD146 (Chemicon International)] for 25 min at 4°C. Appropriate isotype-matched antibodies (BD) were used as negative controls. Data from a minimum of 10,000 viable, 7-AAD-excluding cells were acquired using a Navios® flow cytometer (Beckman Coulter) equipped with three solid-state diode lasers: 405 nm, 40 mW; 488 nm, 22 mW; and 638 nm, 25 mW. List-mode files were analyzed with FlowJo software (Tree Star, Inc.;
Cell morphology of av-MSCs and bv-MSCs
av-MSCs and bv-MSCs were cultured in either DMEM or EGM-2 at low and high passages. Pictures of subconfluent and confluent cells were taken at passages 3 and 6 (n≥2 per cell type).
Immunofluorescence staining of av-MSC and bv-MSC networks in Matrigel
For vWF fluorescence staining of av-MSCs (passage 3) and bv-MSCs (passage 3), cells were seeded separately in a four-well glass chamberslide coated with 250 μL Matrigel per well (750,000 cells/well). After 24 h cells were fixed with dimethyl sulfoxide:methanol (1:4) at 4°C overnight. Then, slides were washed with PBS and treated with Triton X-100 (0.5% in PBS, 7 min at 4°C). After a 90-min blocking step with 3% bovine serum albumin (BSA) in PBS, cells were washed with Tween 20 (3% BSA in PBS) for 5 min. The primary antibody against vWF (Dako; 2.9 μg/mL) was applied overnight at 4°C. Cells were washed twice in PBS, once in PBS with 3% BSA, and once in PBS with 3% BSA and 0.1% Tween 20, followed by incubation with the secondary antibody (Alexa488, goat anti-rabbit, 10 μg/mL) for 1 h at 37°C. Slides were again washed in PBS, mounted with ProLong® Gold antifade reagent (Invitrogen), and analyzed by fluorescent microscopy using an upright microscope system (Leica).
Osteogenic and adipogenic differentiation of av-MSCs and bv-MSCs
To induce osteogenic differentiation, cells were cultured on two-well-chamber slides in DMEM/F12 (Gibco, Invitrogen) supplemented with 0.1 μM dexamethason, 100 μM
For adipogenic induction, cells were grown on two-well-chamber slides in DMEM/F12 supplemented with 10% FBS, 1 μM dexamethason, 200 μM indomethacin, 500 μM 3-isobutyl-1-methylxanthine, and 10 μg/mL insulin (from bovine pancreas; all from Sigma-Aldrich). Control cells were cultured in DMEM/F12 with 10% FBS. To visualize lipid droplets, cells were fixed in 60% 2-propanol and stained with oil red O (stock: 0.5% oil red O in 2-propanol, 3:2 dilution in distilled water) for 10 min.
Sclerotic arteries (ethical approval No. 19-293 ex 07/08) were used as positive controls for alizarin red and oil red O staining.
Lymphocyte proliferation test
Human peripheral blood mononuclear cells (PBMCs) were obtained from heparinized whole-blood samples or buffy coats from healthy subjects using density gradient centrifugation (Axis-Shield) according to the protocol of Magatti et al. [37].
Lymphocyte proliferation was induced either by stimulating “responder” PBMCs (1×105/well in 96-well plate) by the addition of anti-CD3 (Orthoclone OKT3; Janssen-Cilag, final concentration of 12.5 ng/mL) or by coculture with irradiated allogeneic “stimulator” PBMCs [mixed leukocyte reaction (MLR)] at a 1:1 ratio.
To study the effects of bv-MSCs on T-lymphocyte proliferation in cell–cell contact, different amounts of cultured bv-MSCs (1×105, 0.5×105, 0.25×105, 0.125×105, and 0.0625×105) were plated in UltraCulture medium (Lonza) and left to adhere overnight. The next day, bv-MSCs were γ-irradiated (3,000 cGy), and PBMCs with anti-CD3 or PBMCs with allogeneic PBMCs (for MLR) were added to each well, obtaining ratios of “responders” to bv-MSCs of 1:1, 1:0.5, 1:0.25, 1:0.125, and 1:0.0625.
Effect of bv-MSCs on lymphocyte proliferation was also tested by keeping cells separated by a transwell system. Chambers with 0.4-μm-pore-size membranes (Corning) were used to physically separate lymphocytes from bv-MSCs. Different amounts of cultured bv-MSCs (1×105, 0.5×105, 0.25×105, 0.125×105, and 0.0625×105) were plated in UltraCulture medium in the upper chambers of the transwell inserts. The next day, PBMCs with anti-CD3 were added to the lower chambers, obtaining ratios of “responders” to bv-MSCs of 1:1, 1:0.5, 1:0.25, 1:0.125, and 1:0.0625.
Experiments were carried out in triplicates in complete UltraCulture medium, and lymphocyte proliferation was assessed by adding [3H]-thymidine (0.67 μCi/well; PerkinElmer) for 16–18 h after 3 (for PBMC+anti-CD3 stimulation) or 5 (for MLR) days of culture. Cells were then harvested with a Filtermate Harvester (PerkinElmer Life and Analytical Sciences), and thymidine incorporation was measured using a microplate scintillation and luminescence counter (Top Count NXT; PerkinElmer).
Preparation of Cdm
Confluent av-MSCs (passage 1), bv-MSCs (passage 3), and ECs (passage 3) were washed with PBS and then incubated with EGM-2 medium (12 mL per 75 cm2). Control medium (EGM-2) was prepared in parallel in culture flasks without cells. After incubation for 48 h, Cdm and control media were collected and aliquots were stored at −80°C. Prior to application, CdM and control media were thawed and centrifuged at 300 g for 10 min. All cell types were cultivated under standard (37°C, 5% CO2, 21% O2) and reduced oxygen culture conditions (37°C, 5% CO2, 2% O2). Cell numbers were normalized to 105 cells for all subsequent experiments.
Matrigel assay
To investigate whether av-MSCs and bv-MSCs are able to enhance endothelial network formation via paracrine factors, 96-well plates were coated with 40 μL Matrigel per well (BD Biosciences). Confluent ECs were washed with PBS, detached with accutase, and seeded in a density of 104 cells per well. Cells were resuspended with av-MSC-Cdm, bv-MSC-Cdm, EC-Cdm (n≥3 per cell type), or control media obtained from all cell types under two different oxygen concentrations (21% and 2% O2) in triplicates to reach a total volume of 150 μL per well. Network formation was recorded using the cell imaging platform Cell-IQ-Analyzer 2004-01 (Chipman Technologies). Tube length and branching points of endothelial networks were quantified after 12 h of incubation using the AngioJ-Matrigel assay plug-in [20], which was developed and adapted for the ImageJ software (National Institutes of Health) by Diego Guidolin (University of Padova).
Lactate dehydrogenase assay
ECs were seeded in a 96-well plate (6,000 cells per well) and incubated for 72 h with av-MSC-Cdm, bv-MSC-Cdm, EC-Cdm (n≥3 per cell type), and unconditioned control medium obtained at 2% and 21% oxygen. Subsequently, the media were collected and centrifuged for 10 min at 300 g. The supernatants were transferred to a 96-well plate in duplicates and incubated with the reaction mixture provided in the LDH Cytotoxicity Detection Kit (Takara Bio, Inc.) according to the manufacturer's instructions.
Migration assay
The cell migration assay was performed using inserts with 8-μm-pore-sized membranes in a 24-well plate (Cell Biolabs, Inc.). av-MSC-Cdm, bv-MSC-Cdm, and control media obtained at 21% oxygen were placed in triplicates into the wells (n=3 per cell type). ECs were resuspended in serum-free EBM and transferred onto the filter of the insert (300,000 cells/insert). After 4 h of incubation at 37°C, nonmigratory cells were removed from the upper side of the filter. Migratory cells at the bottom side of the filter were stained according to manufacturer's instructions and pictures were taken with an Axio Zoom V16 microscope (Zeiss). Subsequently, migratory cells were removed from the filter with the extraction solution provided in the kit and optical density was measured using a microplate reader (Anthos).
Angiogenic protein analysis
The secretion of angiogenic cytokines/proteins by av-MSCs, bv-MSCs, and ECs was determined using the Raybio Human Angiogenesis Antibody Array C Series 1000 (RayBiotech). This array consists of two membranes, onto which a total of 43 proteins involved in angiogenesis are spotted in duplicates. Pooled Cdm of different isolations of av-MSCs (n=2), bv-MSCs (n=3), ECs (n=3), and non-Cdm (control) obtained at standard oxygen conditions were used for the experiment. Nine hundred and fifty microliters of Cdm was applied per membrane and membranes were processed according to the manufacturer's instructions. Chemiluminescent imaging was performed using the FlourchemQ system and signal densities were analyzed with AlphaView software version 2.0.1.1. (both from AlphaInnotech, Cell Biosciences). Ratios of the respective protein and the internal standard densities were determined, and protein signals from the Cdm were presented as percentage of nonconditioned control media (EGM-2), which was set to 100%.
In addition, quantification of bFGF, human platelet-derived growth factor (PDGF-BB), human placenta growth factor (PlGF), and VEGF was performed using CdM and unconditioned control medium obtained at 21% and 2% oxygen conditions using ELISA (Quantikine Human basic Immunoassay; R&D Systems, Inc.). Cdm of av-MSCs, bv-MSCs, and ECs (n=3 isolations per cell type) was analyzed in duplicates according to the manufacturer's instructions.
Coculture of ECs and MSCs on gelatin
ECs were cocultured with av-MSCs (passage 3, n=2) or bv-MSCs (passage 3, n=2) in duplicates on gelatin-coated chamber slides for 8 days at 37°C (ratio of EC:av/bv-MSC, 2,000:8,000 cells/chamber) under standard oxygen conditions. After harvesting, cells were visualized using immunocytochemistry. Slides were fixed in acetone for 4 min and rehydrated in PBS before immunolabeling for VE-Cadherin (mouse IgG1, 0.3 μg/mL; Santa Cruz) for 30 min at room temperature. Immunoreactivity was visualized with the UltraVision LP Detection System (Thermo Scientific) according to the manufacturer's instructions. Three randomly chosen pictures were used for quantification of total endothelial tube length using ImageJ software (National Institutes of Health).
Coculture of ECs and MSCs on Matrigel
For coculture assays on Matrigel, ECs were seeded in a 96-well plate (104 cells/well) and allowed to form networks for 5 h at 37°C under standard oxygen conditions. av-MSCs (passage 2, n=3) and bv-MSCs (passage 3, n=3) were labeled with CellTracker™ Green CMFDA (1:1,000; Invitrogen) for 30 min at 37°C and added to the endothelial networks in triplicates (ratio of EC:av/bv-MSC, 10,000:3,300 cells/chamber). Cells were observed after 6, 12, and 24 h using the Cell-IQ Analyzer 2004-01 (Chip-Man Technologies).
Statistics
Data are reported as means±standard deviations. Differences in conditioned media among cell types (av-MSCs, bv-MSCs, and ECs) and control were analyzed by one-way analysis of variance with Bonferroni post-hoc corrections. Student's t-test was applied for the quantification of EC tube length in gelatin coculture experiment, for lymphocyte proliferation test, and for ELISA quantification, after testing for normal distribution (Kolmogorov-Smirnov test). Statistical analysis was done using SPSS IBM Statistics 21. A P-value<0.05 was considered as significant.
Results
Phenotypic characterization of av-MSCs and bv-MSCs
Flow cytometric analysis demonstrated that av-MSCs and bv-MSCs showed a similar surface marker profile of common MSC markers. Both cell types were positive for CD90, CD73, CD105, HLA-ABC, CD146, CD63, CD29, CD166, CD13, CD10, and CD49a, and were negative for the immune and EC markers CD45, HLA-DR, CD14, CD3, CD19, CD15, and CD31. In addition, they lacked expression of CD271, AP, and mesenchymal stem-cell like antigen-1 (MSCA-1) (Fig. 1).
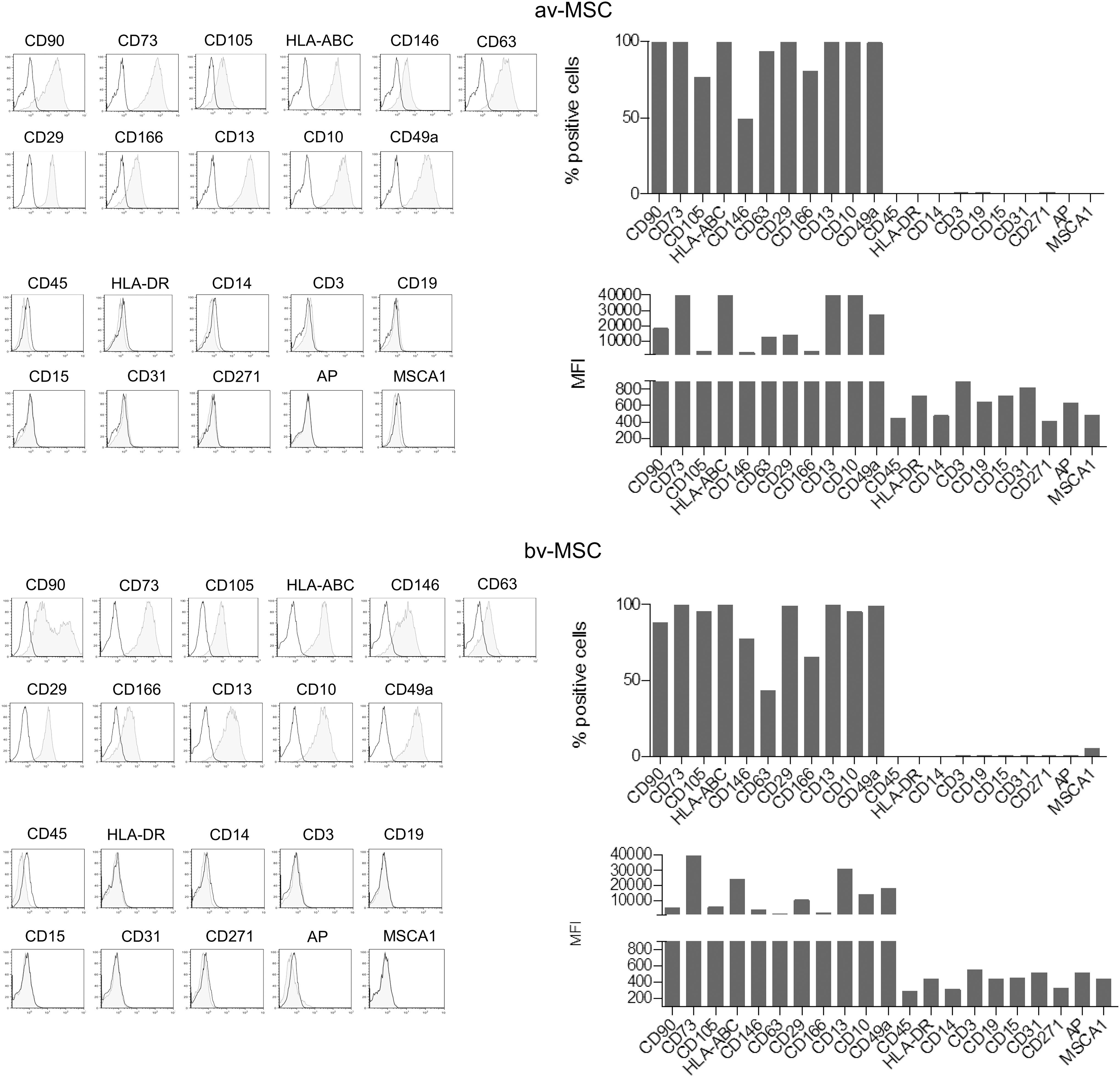
Surface marker profile of av-MSCs and bv-MSCs. Both cell types express CD90, CD73, CD105, HLA-ABC, CD146, CD63, CD29, CD166, CD13, CD10, and CD49a, and lack expression of CD45, HLA-DR, CD14, CD3, CD19, CD15, CD31, CD271, AP, and MSCA-1. Data are representative for at least two different cell isolations per cell type. av-MSCs, amnion-derived avascular mesenchymal stromal cells; bv-MSCs, chorionic-blood-vessel-derived mesenchymal stromal cells; MFI, mean fluorescence intensity; AP, alkaline phosphatase; MSCA-1, mesenchymal stem cell antigen-1.
We reported recently that av-MSCs change their morphology toward a cobblestone-like phenotype when cultured in EGM-2 [14]. Thus, we cultured both MSC types in DMEM and EGM-2. At passage 3, bv-MSCs cultured in EGM-2 showed a fibroblast-like morphology distinct from the cobblestone-like av-MSCs. However, at passage 6, also bv-MSCs adopted a more cobblestone-like morphology at confluence (Fig. 2).
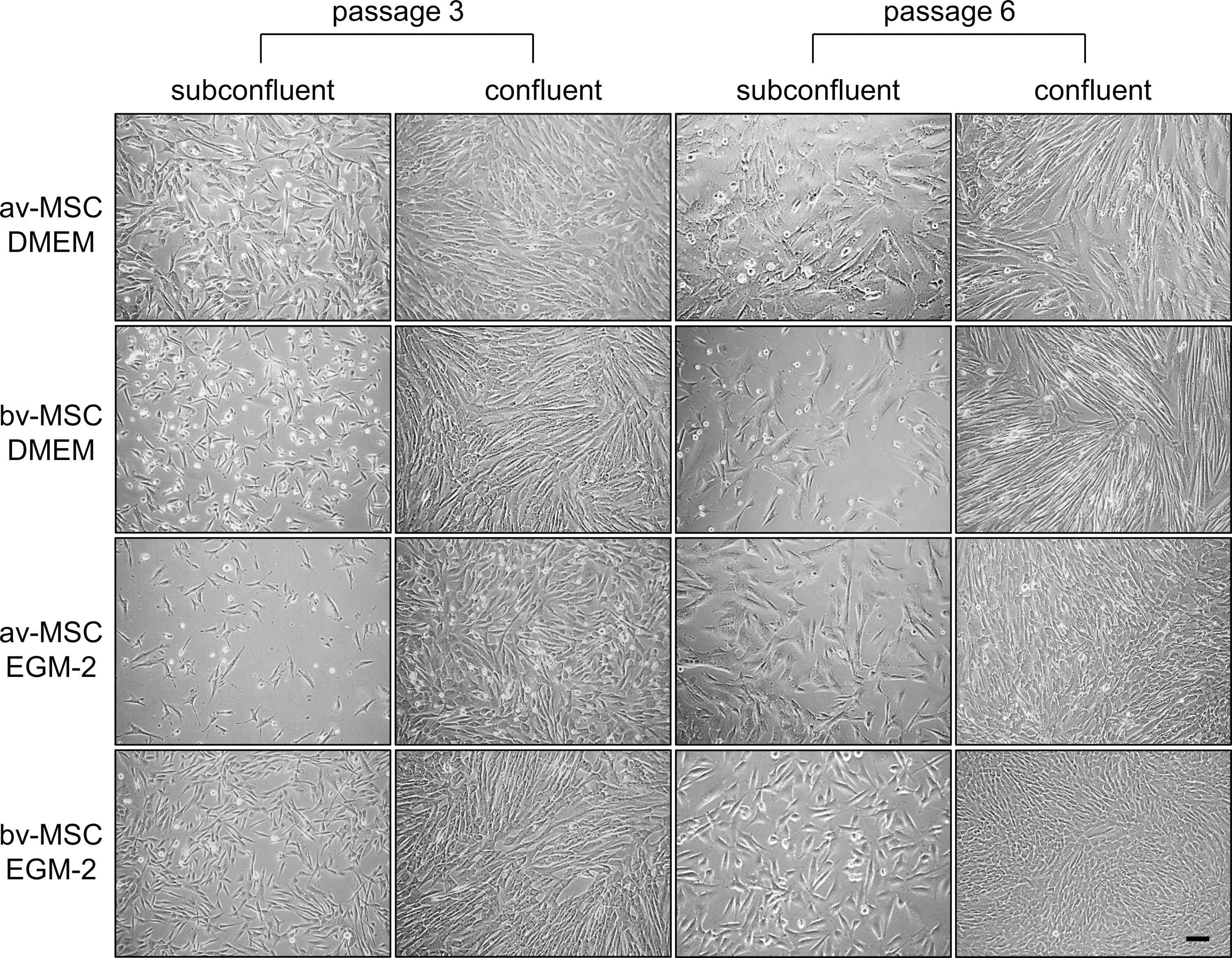
Morphology of av-MSCs and bv-MSCs at low and high passages. av-MSCs and bv-MSCs cultured in DMEM reveal fibroblast-like morphology at confluence. av-MSCs show a cobblestone-like morphology at passages 3 and 6 at confluence when cultured in EGM-2. bv-MSCs cultured in EGM-2 show a fibroblast-like morphology at passage 3 but change their morphology toward a cobblestone-like morphology in passage 6 at confluence. Scale bar=100 μm. DMEM, Dulbecco's modified Eagle's medium; EGM-2, endothelial growth medium-2.
As previously shown, av-MSCs are able to form networks on Matrigel, but still resist differentiation into mature ECs documented by absent staining with the endothelial marker vWF [14]. We now show that also bv-MSCs are able to form network structures on Matrigel that remained negative for vWF (Fig. 3). In addition, both cell types were negative for the muscle cell markers myosin, desmin, and smA (Supplementary Fig. S1; Supplementary Data are available online at
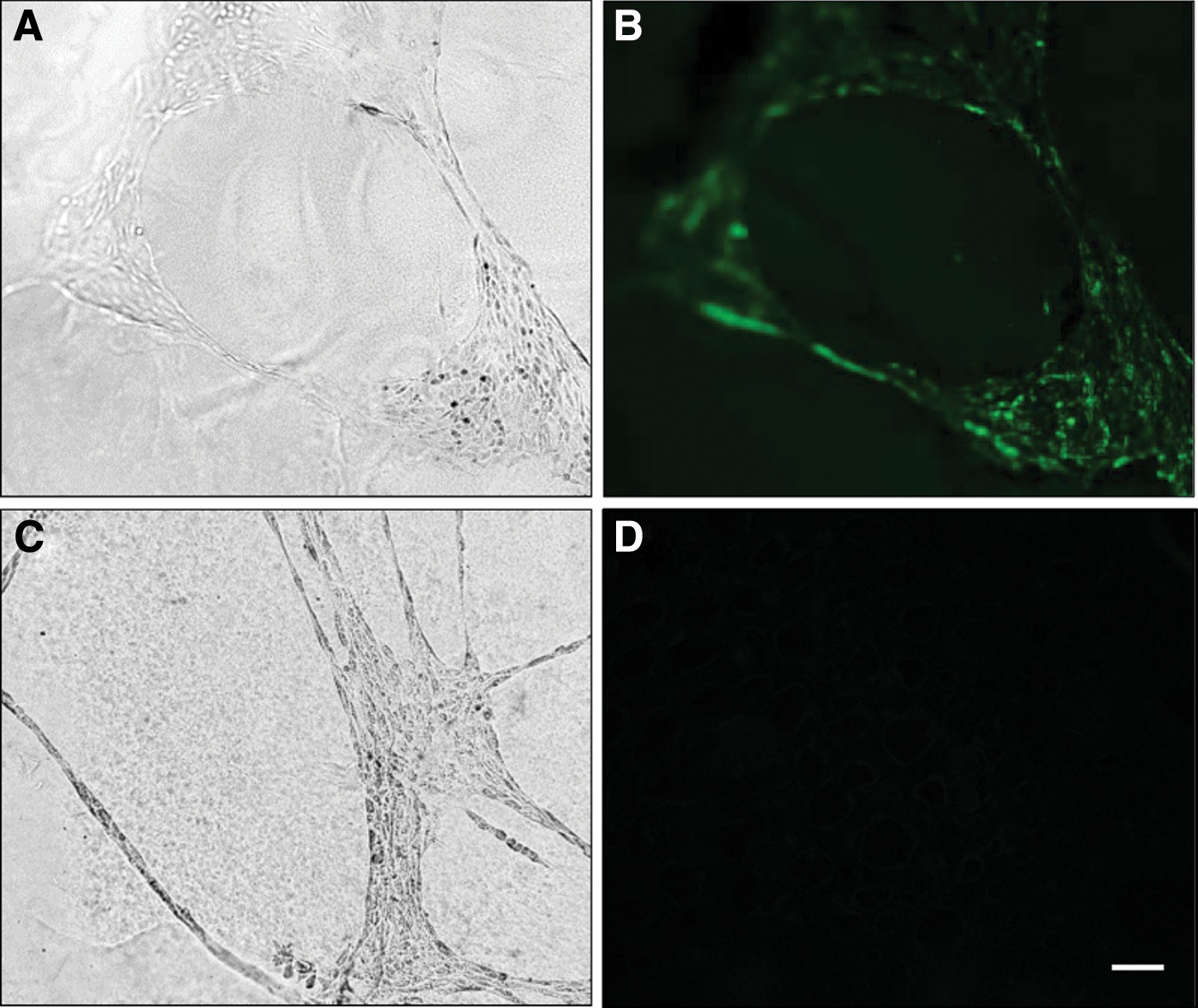
Network formation of ECs and bv-MSCs on Matrigel. Endothelial networks cultured on Matrigel for 24 h [
Osteogenic and adipogenic differentiation potential of av-MSCs and bv-MSCs
Osteogenic induction led to the formation of sparse calcium deposits in bv-MSCs and av-MSCs as shown by staining with alizarin red (Fig. 4A, B). Adipogenic induction led to accumulation of lipid droplets detected with oil red O staining in both cell types (Fig. 4E, F). av-MSCs cultured in DMEM/F12+15% FBS served as undifferentiated negative controls (Fig. 4C, G) and sclerotic arteries as positive controls (Fig. 4D, H) for the respective staining.
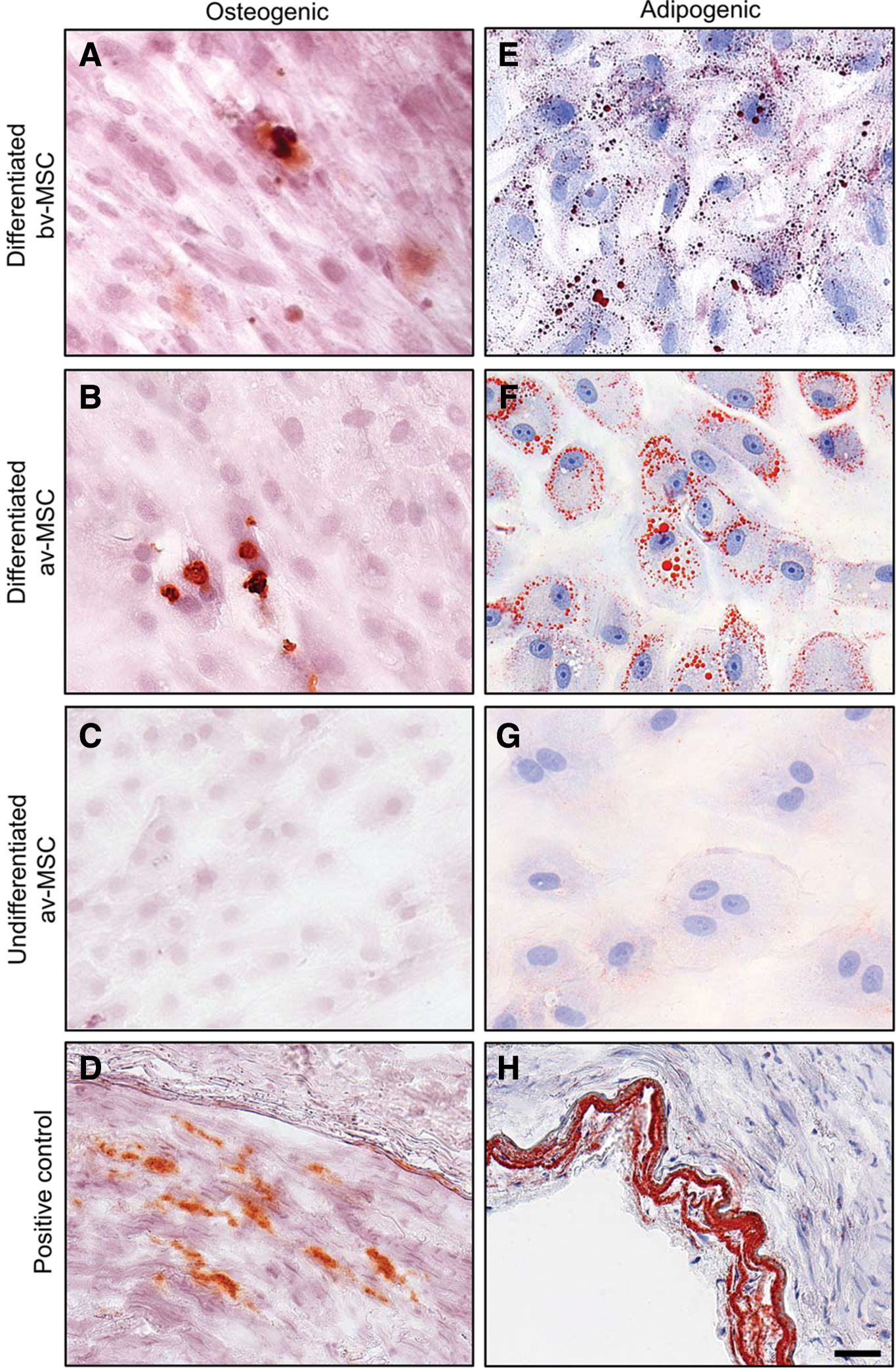
Osteogenic and adipogenic differentiation potential of bv-MSCs and av-MSCs. Cells cultured under osteogenic
Immunomodulatory properties of bv-MSCs
It has been shown that av-MSCs have immunomodulatory properties and are able to inhibit PBMC proliferation induced by CD3 stimulation or by mixed lymphocyte reaction (MLR) [36]. Also, bv-MSCs were able to significantly inhibit PBMC proliferation after activation with anti-CD3, both in direct contact (Fig. 5A) and when physically separated from PBMCs using transwell chambers (Fig. 5B). bv-MSCs also significantly inhibited MLR-induced PBMC proliferation in a direct cell–cell contact setting (Fig. 5C). In all experiments, the inhibitory effects were dose dependent.
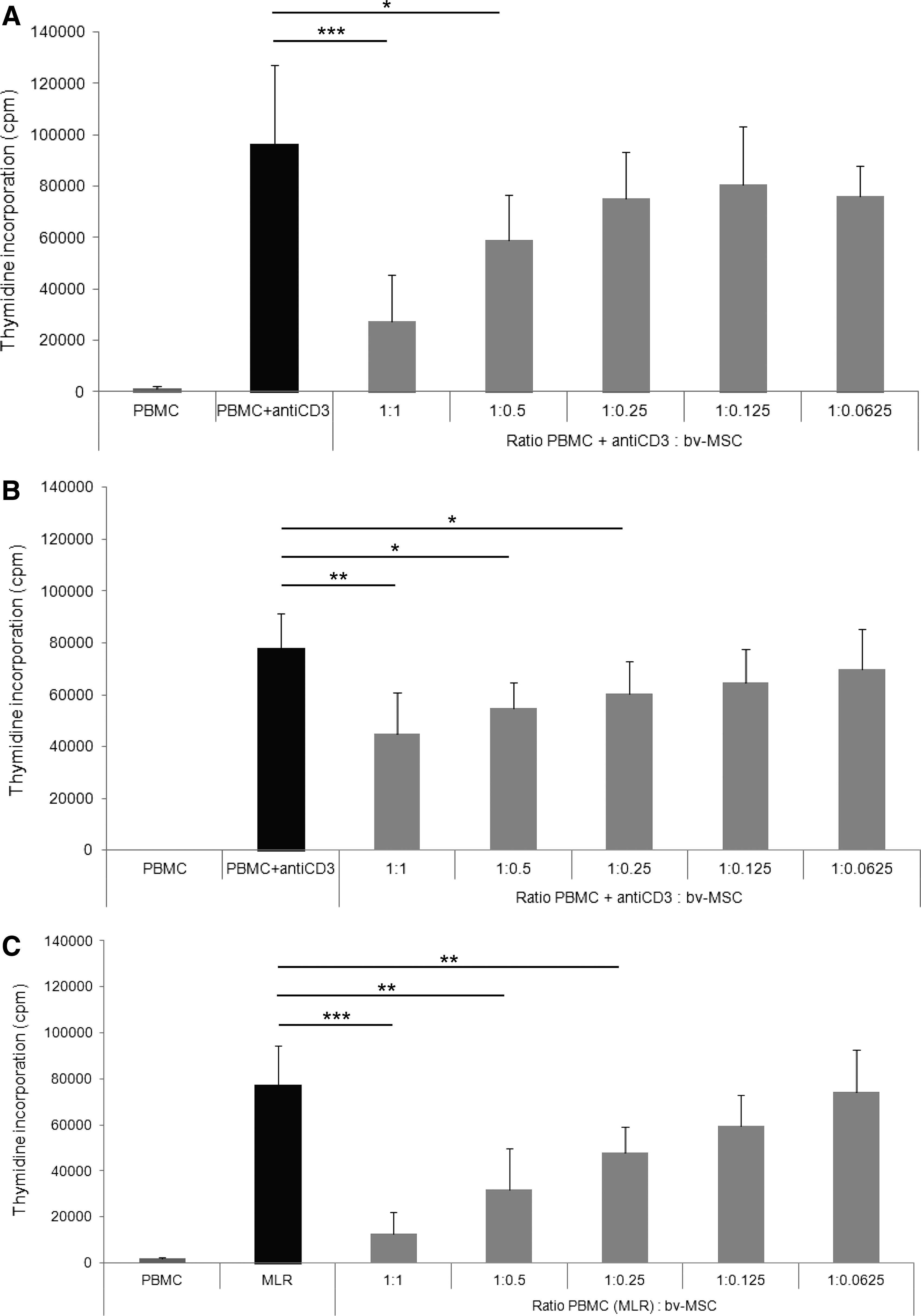
Immunomodulatory properties of bv-MSCs.
Paracrine effects of av-MSC-Cdm and bv-MSC-Cdm on endothelial network formation
av-MSC-Cdm did not increase endothelial network formation compared with unconditioned control medium as measured by endothelial tube length (Fig. 6A, C) or branching points (Fig. 6B, D), independent of the oxygen concentration (21% or 2%). In contrast, bv-MSC-Cdm collected at 21% oxygen significantly increased tube length of endothelial networks (24,270±1,289 μm; control: 19,046±2,948 μm; Fig. 6A) and the number of branching points (302±31; control: 234±46; Fig. 6B) as well as at 2% oxygen (tube length: 25,453±3,322 μm; control: 22,448±1,317 μm; branching points: 340±23; control: 270±59; Fig. 6C, D). No significant differences could be detected between the two oxygen conditions.
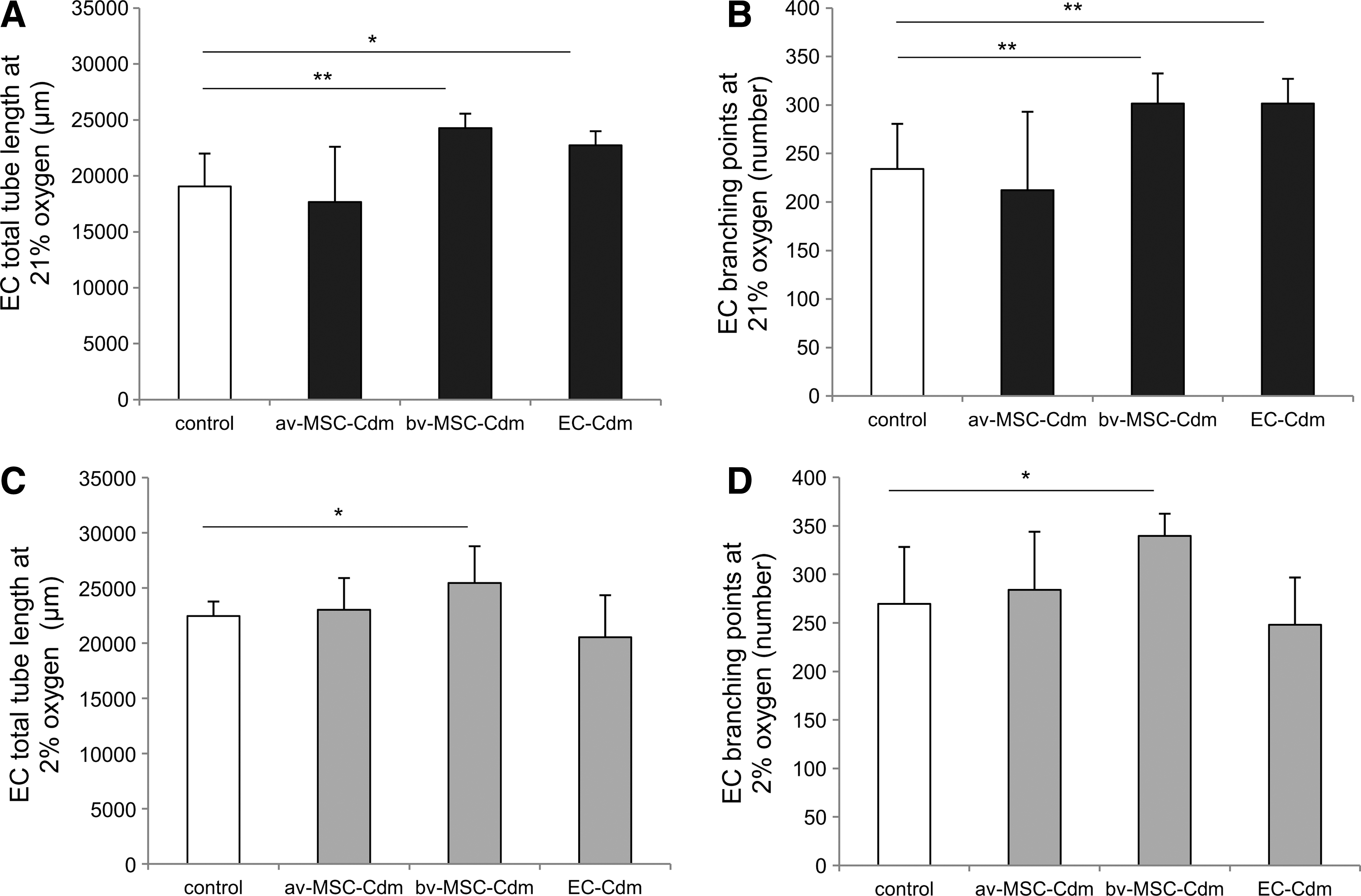
Paracrine effects of av-MSC-Cdm and bv-MSC-Cdm on endothelial tube length and branching points. ECs were incubated with av-MSC-Cdm, bv-MSC-Cdm, EC-Cdm, or unconditioned control medium for 12 h. bv-MSC-Cdm collected at 21% and 2% oxygen stimulated network formation by increasing tube length
EC-Cdm collected at 21% oxygen increased endothelial tube length (22,730±1,261 μm, control: 19,046±2,948 μm) and the number of branching points (302±25; control: 234±46), whereas EC-Cdm obtained at 2% oxygen showed no significant effect (Fig. 6).
Effect of Cdm on EC viability
av-MSC-Cdm obtained at 21% and 2% oxygen significantly decreased lactate dehydrogenase (LDH) activity in the supernatant of EC cultures after 72 h compared with standard control medium (Fig. 7A, 21% oxygen: 58.5%±26.4% of control; Fig. 7B, 2% oxygen: 46.2%±17.5%). bv-MSC-Cdm showed similar results (21% oxygen: 58.9%±16.8%; 2% oxygen: 50.9%±6.7%). As LDH is an enzyme that is released into the culture medium by damaged cells [21], these results showed that both av-MSCs and bv-MSCs clearly enhanced viability of ECs under standard culture conditions. EC-Cdm had a less-pronounced effect on cell viability, but still significantly reduced LDH activity to 70.9%±8.3% (21% oxygen) and 58.9%±19.6% (2% oxygen) of control.
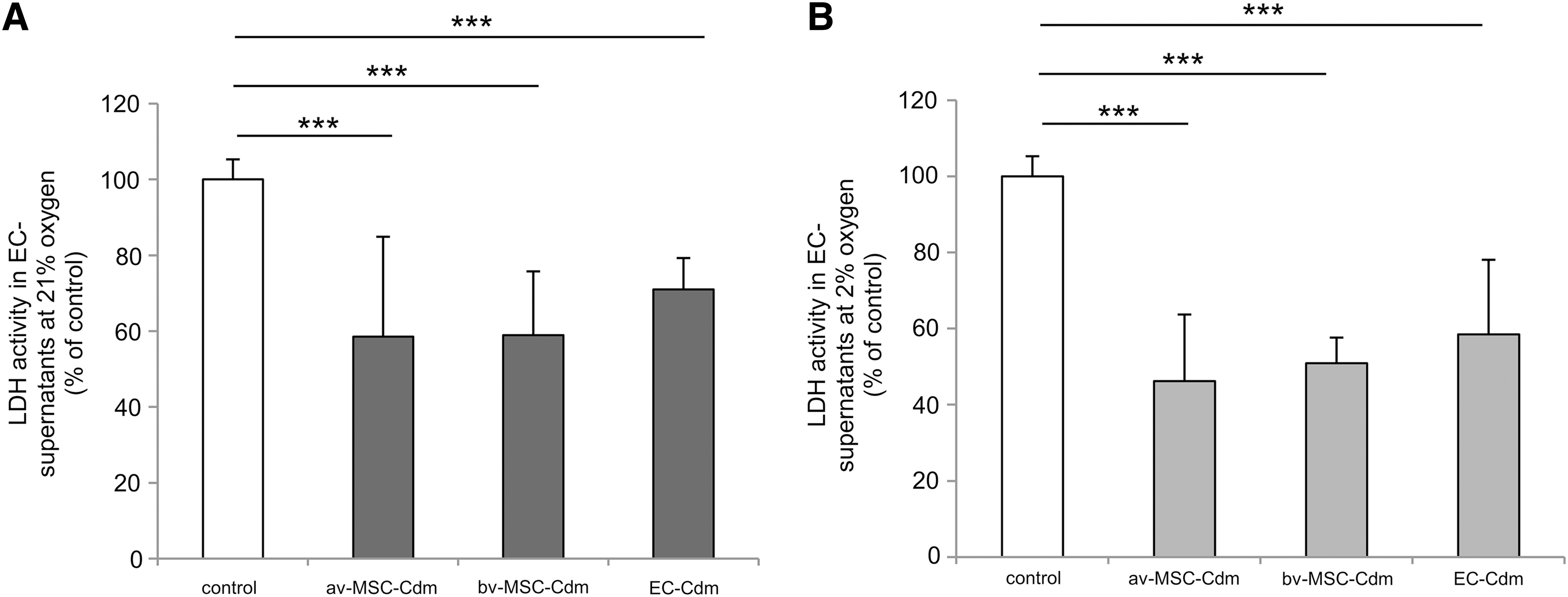
Effect of Cdm on EC viability. av-MSC-Cdm, bv-MSC-Cdm, and EC-Cdm significantly reduced LDH activity in EC culture supernatants compared with control media (set to 100%). Cdm was obtained at 21%
Effect of Cdm on cell migration
Significantly more ECs migrated toward bv-MSC-Cdm (165%±24% of control) than toward av-MSC-Cdm (115%±34% of control) (Fig. 8). Migration toward standard control medium was set to 100%. Effects of av-MSC-Cdm did not differ significantly from control medium.
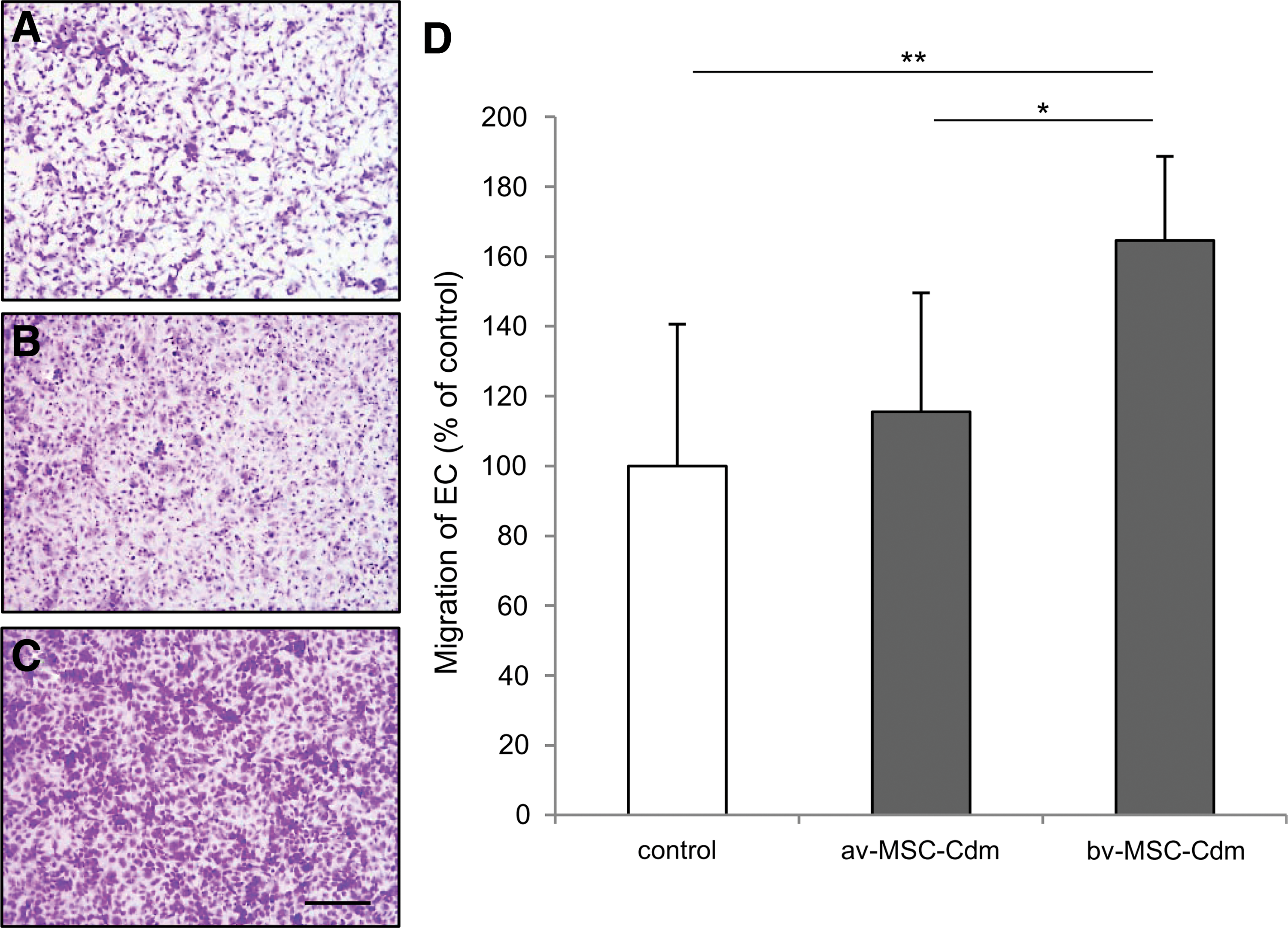
Effect of Cdm on EC migration. ECs were seeded into 8-μm-pore-sized membranes and allowed to migrate toward av-MSC-Cdm and bv-MSC-Cdm. Representative pictures of EC migration toward control media
Detection of angiogenic proteins secreted by av-MSCs, bv-MSCs, and ECs
Compared with control media (set to 100%), bv-MSC-Cdm contained elevated levels of the proangiogenic proteins angiogenin (108%±28%), angiopoietin-1 (49%±3%), angiopoietin-2 (36%±7%), CXC-chemokine GRO (161%±10%), interleukin-6 (IL-6, 129%±41%), IL-8 (169%±60%), monocyte chemoattractant protein 1 (MCP-1, 72%±3%), thrombopoietin (22%±0.2%), angiopoietin receptor Tie-2 (36%±8%), urokinase receptor (30%±3%), and VEGF (30%±2%) (Fig. 9A, B). Also, tissue inhibitor of metalloproteinases-1 and -2 (TIMP-1, 124%±13% and TIMP-2, 74%±3%) and tumor necrosis factor-α (TNF-α, 60%±8%) were significantly higher compared with control. Further, bv-MSCs secreted the angiogenesis inhibitors angiostatin (26%±8%) and endostatin (47%±5%). av-MSC-Cdm showed a similar pattern but mostly with weaker signals of the detected factors. In contrast to bv-MSCs, av-MSCs secreted interferon-γ (IFN-γ, 12%±4%) and RANTES (also known as C–C motif chemokine 5, 22%±7%). av-MSCs did not secrete angiopoietins-1 and -2, angiostatin, endostatin, MCP-1, thrombopoietin, Tie-2, TNF-α, and VEGF. Compared with control media, EC-Cdm contained high amounts of angiopoietin-2 (260%±23%), GRO (616%±178%), IL-8 (488%±111%), and MCP-1 (436%±44%).
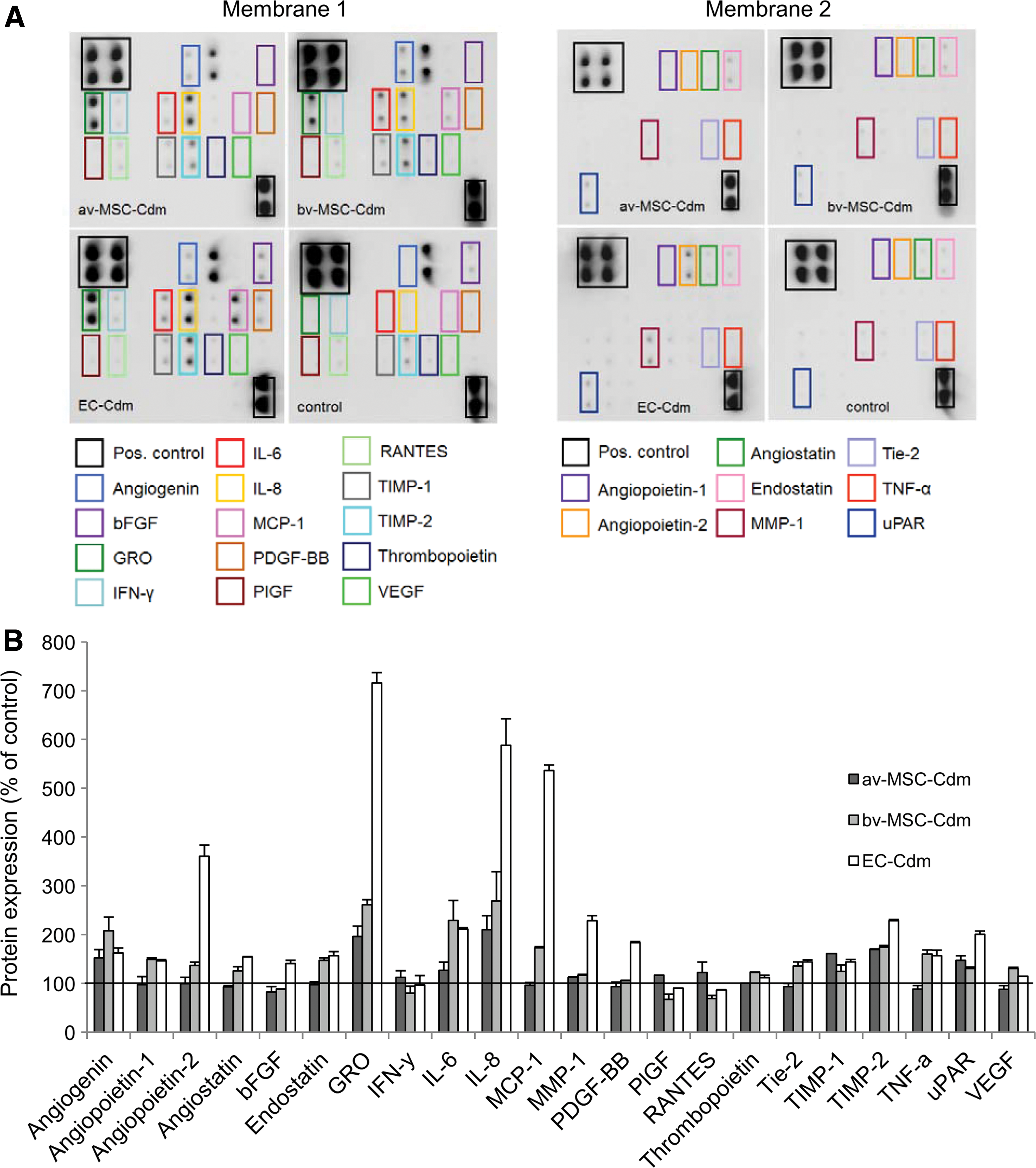
Secretion of angiogenic proteins by av-MSCs, bv-MSCs, and ECs at 21% oxygen.
Quantification of bFGF, PDGF-BB, PlGF, and VEGF secretion by ELISA
The concentrations of bFGF, PDGF-BB, PlGF, and VEGF in the Cdm were further quantified using ELISA (Fig. 10A: 21% oxygen, B: 2% oxygen). In Cdm collected from av-MSCs, lower values of bFGF and VEGF were found in av-MSC-Cdm compared with control medium. bv-MSCs secreted VEGF at 21% and 2% oxygen (1,538.5±693.4 pg/mL and 2,485.4±552.5 pg/mL, respectively), while lower amounts of bFGF where found in bv-MSC-Cdm compared with control medium. No significant difference was detected between the two oxygen conditions. ECs showed secretion of bFGF, PDGF-BB, and PlGF and reduced amounts of VEGF in Cdm under both oxygen conditions.
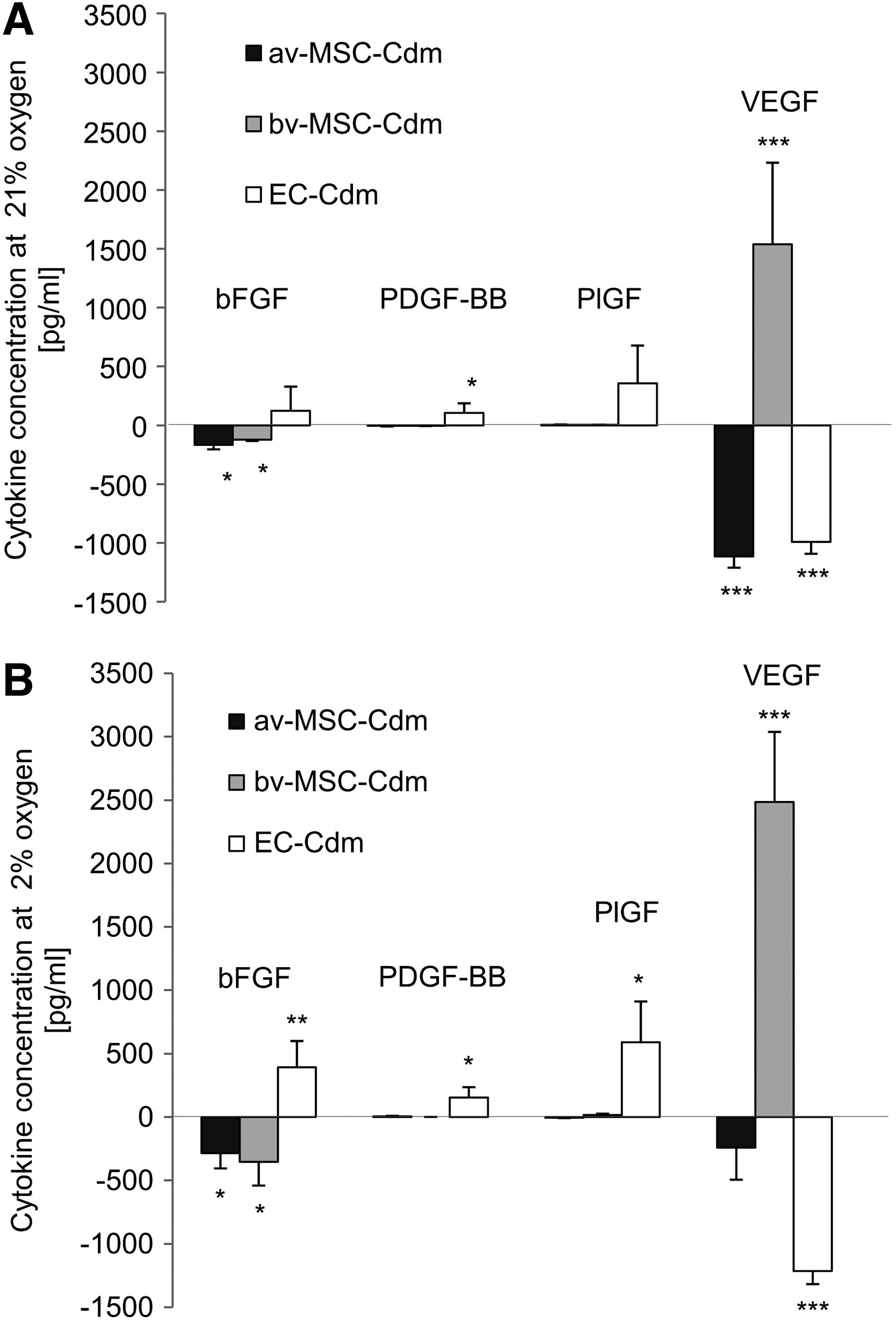
Quantification of bFGF, PDGF-BB, PlGF, and VEGF in Cdm obtained at 21%
Coculture of ECs and MSCs on gelatin
Direct coculture of ECs and av-MSCs or bv-MSCs on normal gelatin-coated tissue culture flasks resulted in the formation of vessel-like structures by the ECs (Fig. 11A, B: ECs stained red with VE-Cadherin). The length of the formed EC alignments was significantly higher in cocultures with bv-MSCs (total length of vessel-like structures 12,232 μm±2,831 μm) than with av-MSCs (6,680 μm±423 μm) (Fig. 11C).
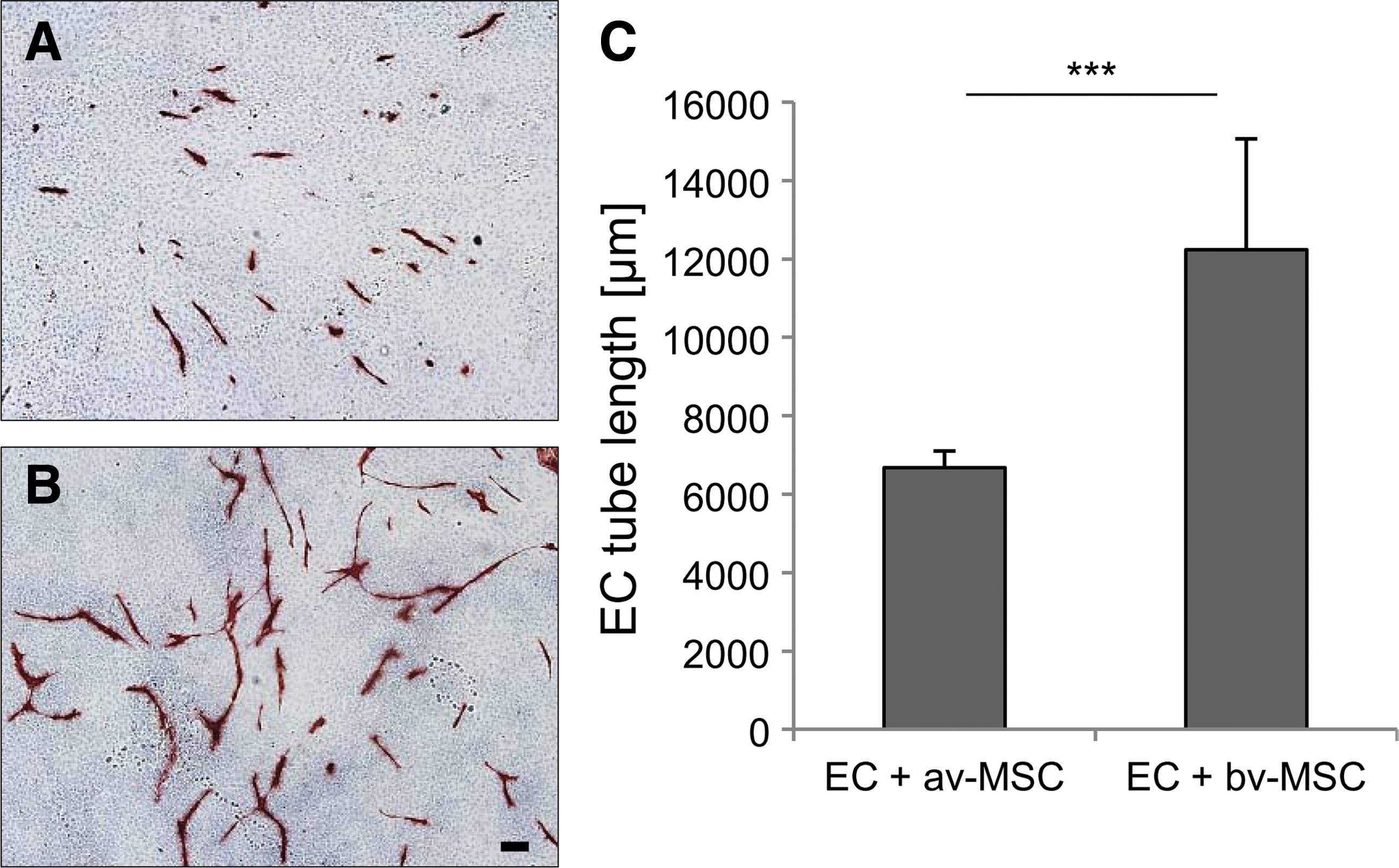
Coculture of ECs with av-MSCs and bv-MSCs on gelatin. Representative pictures of ECs cocultured with av-MSCs
Coculture of ECs and MSCs on Matrigel
In the Matrigel assay both av-MSCs and bv-MSCs incorporated into pre-existing endothelial networks and stabilized them (Fig. 12). MSCs were labeled with green fluorescence and added to the endothelial networks (Fig. 12A–C) after 5 h. As has already been shown for av-MSCs [14], bv-MSCs within these networks remained negative for vWF (Fig. 3).
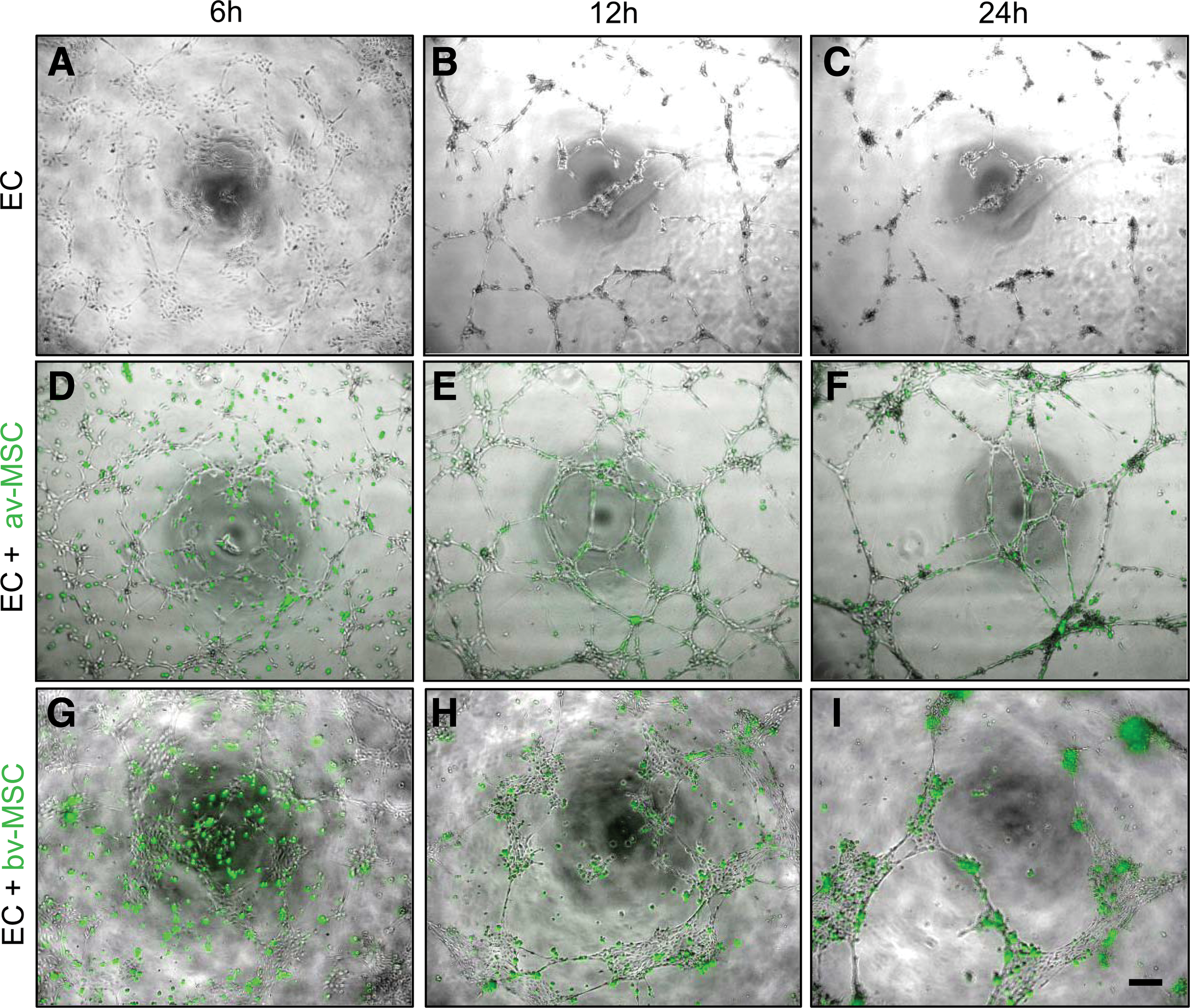
Coculture of ECs with av-MSCs and bv-MSCs on Matrigel. ECs were seeded on Matrigel and allowed to form networks
Discussion
In this study, we evaluated the angiogenic effect of MSCs isolated from two different niches within the same organ (placenta): av-MSCs derived from the avascular amnion and perivascular bv-MSCs derived from chorionic vessel outgrowths.
While av-MSCs have been extensively investigated, information about bv-MSCs is rare. We therefore characterized bv-MSCs in comparison to av-MSCs according to criteria suggested by the International Society of Cellular Therapy [22]. MSCs are commonly identified via the expression of specific surface markers, including CD73, CD105, and CD90 and lack the expression of various immune cell and ECs markers, such as CD45, CD14, HLA-DR, and CD34 [2,22]. In our analysis we included additional markers known to be expressed by MSCs [23,24]. bv-MSCs express most of these markers and have a very similar surface marker profile to av-MSCs. Both cell types lack expression of MSCA-1 and CD271, which have been suggested as markers for an especially clonogenic subpopulation of bone marrow MSCs [24 –27]. While MSCA-1 and AP are known to be only expressed by bone-marrow-derived MSCs [24], the expression of CD271 has been described in a small subpopulation of av-MSCs before [17].
Further, we show that bv-MSCs can be differentiated toward the adipogenic and osteogenic lineages in vitro. Even though this differentiation potential is a common MSC characteristic [22], its role in in vivo transplantation settings is still unclear, as generally only a few differentiated cells can be detected in the treated tissues. Instead, it is now widely recognized that the cytoprotective effects of MSCs can to a considerable extent be ascribed to the secretion of soluble factors [28,29]. Cytokines and other factors are assumed to have a beneficial effect on the injured tissue and/or on a present immune reaction.
The low immunogenicity of MSCs allows an allogenic application [2]. A large number of clinical trials show that ex-vivo-expanded MSCs can be safely administered without immune reactions in the recipient [30,31]. MSCs have been successfully applied to improve the engraftment of hematopoietic stem cells after transplantation [32], for the treatment of graft-versus-host disease [33], and osteogenesis imperfecta [30,34].
Many studies show that also av-MSCs possess beneficial immunomodulatory properties. After transplantation into neonatal swine and rats, human av-MSCs were able to engraft without immunosuppression [35]. Human- and mouse-derived av-MSCs improved bleomycin-induced lung fibrosis in a mouse model [36]. In vitro, av-MSCs suppressed T-lymphocyte proliferation [37,38], and prevented differentiation of monocytes into dendritic cells [39].
av-MSCs are also known to secrete a variety of immunomodulatory factors [13,37,39 –42]. The application of Cdm from av-MSCs reduced progression of lung fibrosis in a mouse model [43] and led to favorable clinical outcomes when treating tendon and ligament injuries in horses [44].
In our study, we could demonstrate that bv-MSCs possess immunosuppressive properties similar to av-MSCs [37,38]. bv-MSCs inhibited the proliferation of activated PBMCs both in direct cell-to-cell contact as well as via soluble factors when separated by transwell chambers, suggesting that bv-MSCs are suitable for a use in cell transplantation settings.
We further demonstrated that both av-MSCs and bv-MSCs secrete paracrine factors that promote EC viability, as shown by reduced LDH secretion of ECs cultured in av-MSC-Cdm or bv-MSC-Cdm. The effect of Cdm was significant even though ECs were cultured under standard conditions, without any evidence that their proliferation was altered or that cells were undergoing increased apoptosis or necrosis. In future studies, it will be interesting to investigate the pathways leading to reduced LDH release under the influence of av-MSC-Cdm and bv-MSC-Cdm.
While the effect on EC viability was equally good for both cell types, Cdm from bv-MSCs had a more pronounced effect on EC migration and network formation. A reason for this could be that bv-MSCs, in contrast to av-MSCs, secreted noticeable amounts of VEGF, an important inducer of angiogenesis, as shown by quantitative ELISA analysis. While MSCs from bone marrow are known to secrete VEGF [45 –48], its secretion by av-MSCs has not been reported before, which is in accordance with our results. As less VEGF was found in the Cdm of av-MSCs compared with the control medium, we speculate that these cells take up VEGF from the medium and metabolize it in a similar extent as ECs.
We applied a semiquantitative angiogenesis protein array to analyze the Cdm in order to detect further proteins that could be responsible for an angiogenesis-promoting effect apart from VEGF. Both av-MSCs and bv-MSCs expressed angiogenin. This protein was first isolated from the supernatant of the human adenocarcinoma cell line HT29 and effectively stimulated angiogenesis in in vivo assays [49]. Angiogenin acts via its ribonuclease activity, degradation of basement membrane, and activation of angiogenic signaling transduction [50]. We further found strong signals for GRO, which detects CXCL1, CXCL2, and CXCL3. These are part of the CXC chemokine family and possess angiogenic properties [51,52]. IL-8 (also known as CXCL-8) is part of this family as well, and high amounts were expressed by both av-MSCs and bv-MSCs. IL-8 directly enhances EC survival, proliferation, and capillary endothelial tube formation [53,54]. Especially bv-MSCs secreted high levels of IL-6, which has been shown to stimulate proliferation and migration of ECs as well as tube formation of blood-derived endothelial progenitor cells in vitro [55 –57]. Further, TIMPs-1 and -2 were secreted by av-MSCs and bv-MSCs. Matrix metalloproteinases play an important role in angiogenesis by degrading extracellular matrix, and their inhibitors TIMPs-1 and -2 are involved in different stages during this process [58]. The differences between av-MSCs and bv-MSCs regarding the amount of VEGF secretion were less pronounced using this semiquantitative antibody array as compared to the ELISA. However, the tendency was similar, showing a higher secretion by bv-MSCs compared with av-MSCs.
Interestingly, also ECs secreted high levels of some of the proangiogenic proteins, which could explain the viability-enhancing effect of EC-Cdm as described previously.
The secretion of most of these cytokines by the amnion and by isolated av-MSCs has been reported before, and they have been proposed to contribute to the beneficial effect of the amnion in wound healing [59]. As bv-MSCs seem to secrete even higher amounts of these cytokines, these cells might be a promising alternative source. For the collection of Cdm, av-MSCs from passage 1 and bv-MSCs from passage 3 were used. This was due to the large amount of av-MSCs available already at this early passage number and the observation that the viability and proliferation of these cells is better at earlier than at higher passages. Interestingly, bv-MSC-Cdm still seemed to be more effective than av-MSC-Cdm in spite of the fact that it was collected at a higher passage. In future studies, it would be of great importance to test the functionality of both cell types at higher passages in case they need to be expanded ex vivo in larger scale for clinical applications. In addition, it needs to be taken into account that angiogenesis is a complex process, and that it remains a challenge to determine the interaction of the factors involved. Controversy exists, among others, on the angiogenic actions of certain proteins, including TNF-α and IFN-γ, which have been ascribed both pro- as well as antiangiogenic effects [60,61].
Low oxygen concentrations are known to promote angiogenic processes induced by bone-marrow-derived MSCs [4,16]. Culture of av-MSCs and bv-MSCs at 2% oxygen did not result in enhanced endothelial network formation or EC viability compared with 21%. The secretion of VEGF by bv-MSCs was increased at 2% oxygen, while in av-MSC-Cdm less VEGF was detected under these conditions.
Interestingly, at 21% oxygen EC-Cdm resulted in a significantly increased endothelial network formation in the Matrigel assay, whereas the effect of EC-Cdm generated at 2% oxygen was similar to unconditioned control medium. This raises the questions whether MSCs surrounding ECs are needed to stimulate endothelial outgrowth and hence angiogenesis in a low-oxygen environment.
To identify the effects of direct cell-to-cell contact, we performed cocultures of ECs and av-MSCs or bv-MSCs. Again, bv-MSCs showed a stronger proangiogenic effect, as EC alignments were significantly longer in cocultures with bv-MSCs than in the ones with av-MSCs. In the Matrigel assay, both cell types were able to adhere to and support endothelial network formation without differentiation into ECs as documented by absent expression of the endothelial marker vWf. Thus, even though MSCs do not seem to be able to take over the functional role of ECs, several studies have shown that the coapplication of MSCs with endothelial or endothelial progenitor cells is required for the formation of stable, functional vessels in vivo [48,62 –64]. Melero-Martin et al. and Au et al. demonstrated a perivascular location of MSCs in the newly formed vessels, suggesting that MSCs act as pericytes [48,62]. Pericytes are mural cells that closely encircle ECs of arterioles, venules, and capillaries, while larger vessels are usually surrounded by smooth muscle cells. They play an essential role in the stabilization of vessels and are involved in angiogenesis and vasculogenesis [65]. In situ, MSCs are often located in direct vicinity of blood vessels, and it has been hypothesized that either the so-called MSCs are actually pericytes or that pericytes are a type of MSCs [25,66,67]. However, the identity and characterization of pericytes (and MSCs) is still not reliably resolved and controversially discussed [66,67]. In addition to a vessel-stabilizing effect, Hofmann et al. showed that MSCs (from cord blood) contribute to in vivo neovasculogenesis via oxygen sensing [64].
Recently, we have demonstrated positive effects of av-MSCs on capillary formation in a mouse model in vivo [68]. In a subsequent in vivo study it is planned to compare both subsets of cells with respect to capillary formation and wound healing to support the hypothesis of this article. It has been shown that MSCs derived from different sources regulate angiogenesis via distinct mechanisms and that their identity also influences the functional properties of in vivo vessel implants [69,70]. In our study, we investigated MSCs isolated from a perivascular as well as an avascular niche, and could show that both have angiogenic properties. However, bv-MSCs, which are closely located to blood vessels in situ, were more potent in inducing angiogenesis, as shown by a significant effect on endothelial network formation. This is probably the result of a higher secretion of angiogenic cytokines, especially of VEGF.
With regard to a therapeutic treatment, bv-MSCs might be valuable to stimulate angiogenesis especially in ischemic tissues. av-MSCs, however, could be beneficial in conditions when it is required to promote the survival and stabilization of blood vessels without the risk of unmeant angiogenesis. In a previous study we could show that av-MSCs resist differentiation into mature ECs by upregulation of antiangiogenic genes and proteins [16]. This seems to be in accordance with the characteristic of the amnionic membrane to remain avascular even though being located next to the highly vascularized chorion.
The advantage of both of these placenta-derived MSCs is that they are available in large supply and can be isolated noninvasively. They are immunologically well tolerated, do not form tumors in the recipient and are cells of fetal origin [14,71]. The combination of their immunomodulatory and EC-supporting properties makes them promising tools for blood vessel reconstruction.
Funding
This work was supported by the Franz-Lanyar Foundation (project no. 363). Gregor Weiss was supported by the Austrian Science Fund (FWF, project P24739).
Footnotes
Acknowledgments
The authors thank the research nurse Bettina Amtmann of the Clinic of Obstetrics and Gynecology for placenta collection, and Kerstin Hingerl, Rudolf Schmied, and Monika Siwetz from the Institute of Cell Biology, Histology, and Embryology, Medical University of Graz, Austria, for their valuable technical assistance and expertise and Julia Kröpfl of the Department of Health Sciences and Technology, ETH Zürich, for the help with statistical issues.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.