
Abstract
Mesenchymal stem cells (MSCs) are being tested in a wide range of human diseases; however, loss of potency and inconsistent quality severely limit their use. To overcome these issues, we have utilized a developmental precursor called the hemangioblast as an intermediate cell type in the derivation of a highly potent and replenishable population of MSCs from human embryonic stem cells (hESCs). This method circumvents the need for labor-intensive hand-picking, scraping, and sorting that other hESC-MSC derivation methods require. Moreover, unlike previous reports on hESC-MSCs, we have systematically evaluated their immunomodulatory properties and in vivo potency. As expected, they dynamically secrete a range of bioactive factors, display enzymatic activity, and suppress T-cell proliferation that is induced by either allogeneic cells or mitogenic stimuli. However, they also display unique immunophenotypic properties, as well as a smaller size and >30,000-fold proliferative capacity than bone marrow-derived MSCs. In addition, this is the first report which demonstrates that hESC-MSCs can inhibit CD83 up-regulation and IL-12p70 secretion from dendritic cells and enhance regulatory T-cell populations induced by interleukin 2 (IL-2). This is also the first report which shows that hESC-MSCs have therapeutic efficacy in two different autoimmune disorder models, including a marked increase in survival of lupus-prone mice and a reduction of symptoms in an autoimmune model of uveitis. Our data suggest that this novel and therapeutically active population of MSCs could overcome many of the obstacles that plague the use of MSCs in regenerative medicine and serve as a scalable alternative to current MSC sources.
Introduction
M
MSCs home to injured/inflamed tissue and are thought to provide therapeutic support through a multifaceted mechanism. They secrete a dynamic assortment of bioactive cytokines, trophic factors, and anti-inflammatory molecules such as transforming growth factor beta (TGFβ) [5], monocyte chemotactic protein 1/chemokine (C-C motif ) ligand 2 (MCP-1/CCL2) [6], indoleamine 2,3-dioxygenase (IDO) [7], prostaglandin E2 (PGE2) [8], heme oxygenase-1 (HO-1) [9], monokine induced by gamma interferon (MIG), and interferon gamma (IFNγ)-inducible protein 10 (IP-10) [10] in response to environmental cues. In addition to paracrine-acting factors, MSCs use direct cell-to-cell contact [3,11,12] and influence the activities of different immune cell populations [3,13 –19]. They may also recruit and/or activate endogenous progenitors to facilitate tissue repair at injury sites [20,21]. Differentiation and long-term engraftment may be another mechanism by which MSCs contribute to tissue repair, yet this is thought to play a relatively minor role in their therapeutic activity [22,23].
BM has been the most commonly used source for MSCs in both preclinical animal models and clinical trials currently underway. However, differences in quality of donor cells, tissue sources, and culture methods have caused numerous inconsistencies in the reported in vivo effectiveness of MSCs [24 –27]. Their limited quantity necessitates in vitro culture and expansion in order to obtain sufficient numbers for clinical use; however, this process may also negatively influence in vivo potency and lead to further inconsistencies [28 –31]. The use of human embryonic stem cells (hESCs) or induced PSCs as a source for MSCs may help circumvent many of these issues. Given their pluripotent nature, hESC-derived MSCs can be used to generate unlimited amounts of early-passage MSCs of a consistent quality; while donor-dependent adult tissue sources are non-replenishable, subject to variable quality, and need to be screened for pathogens with every new donor. Moreover, hESC-MSCs can provide corrective functions for diseased, defective, or old MSCs that autologous MSC therapy from a diseased patient cannot provide [32,33].
Since 2005, several groups have reported the derivation of MSCs from PSCs using a variety of methods. These include OP9 co-culture [34,35], embryoid body (EB) formation [36], direct plating of hESCs into MSC media [37 –39] with cell sorting [40,41], scraping [42], or manual picking and expansion of “raclures,” the differentiated areas that spontaneously arise on the edges of hESC colonies [43,44]. More recent studies have used inhibitors of TGFβ and MAPK signaling pathways to augment differentiation of MSCs from hESCs or EBs [45 –47]. In vitro characterization shows that hESC-derived MSCs express a host of typical MSC cell surface markers (CD73, CD90, CD105, CD44, etc.) [38,39,41,48], and they have the ability to differentiate into bone, fat, and cartilage [34,37,42], similar to other sources of MSCs. Gene expression profiling has been used to compare hESC-MSCs with those derived from adult BM [34,40] and while some differences exist, the findings suggest that the two types of MSCs are quite similar, supporting the use of hESCs as an alternative source for generating MSCs for clinical use.
While the feasibility of generating hESC-MSCs has been shown with a variety of methods, their ability to sense and respond to environmental cues, to impact immune cell function, and to function therapeutically in vivo have not been explored in as much detail as they have been for BM-MSCs. In this study, we describe a novel and efficient method for generating hESC-MSCs that obviates the need for scraping, sorting, or hand-picking cells and takes advantage of a versatile precursor cell called the hemangioblast to bridge the differentiation process between PSCs and multipotent MSCs. A battery of in vitro and in vivo assays confirms that hESC-MSCs have the capacity to respond to environmental cues, influence immune cell function, and exert therapeutic effects which are strong enough to reduce clinical symptoms in two different autoimmune disease models.
Materials and Methods
hESC culture and differentiation to MSCs
Human ES cells (MA09, derived from blastomeres, as previously described [49,50]) were cultured in Knockout DMEM, 20% serum replacement (Gibco, Grand Island, NY), 10 ng/mL basic fibroblast growth factor (bFGF), 60 μM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO), penicillin/streptomycin, GlutaMAX, and non-essential amino acids and passaged with 0.05% trypsin-EDTA (Life Technologies, Carlsbad, CA) or in Primate ES Cell Medium (Reprocell, Kanagawa, Japan)+10 ng/mL bFGF according to the manufacturer's instructions. hESC cultures were maintained on irradiated mouse embryonic fibroblasts (GlobalStem, Inc., Rockville, MD) or on Matrigel (BD Bioscience, San Jose, CA) by using mTeSR medium (Stem Cell Technologies, Vancouver, Canada). For EB formation, hESCs were plated on low adherence plates (Corning, Corning, NY) in Stemline II (Sigma-Aldrich) plus BMP4 and vascular endothelial growth factor (VEGF) (50 ng/mL) for 4 days; bFGF (22.5 ng/nL) was added for the last 2 days. EBs were dissociated with 0.05% trypsin-EDTA and single cells were replated in Methocult H4536 (Stem Cell Technologies), with 1×Excyte (Millipore, Billerica, MA), VEGF, Flt3-ligand (FL), thrombopoietin (50 ng/mL each), and bFGF (30 ng/mL) to enable hemangioblast formation, as previously described [51,52]. BMP4 was from Humanzyme (Chicago, IL); all other cytokines in this study were from Peprotech (Rocky Hill, NJ), unless otherwise noted. After 7–12 days, cultures were harvested, rinsed extensively with 1×Dulbecco's phosphate-buffered saline (PBS) (Life Technologies), and plated as p0 in MSC growth conditions, as described later.
MSC culture and flow cytometry analysis
hESC-MSCs were grown in MSC growth medium (αMEM+16%–20% Hyclone fetal calf serum; Thermo-Fisher, Waltham, MA) on Matrigel-coated plates until passage 3/4 (p3/4) when matrigel was no longer used. MSCs were passaged using 0.05% trypsin-EDTA or TrypLE (Life Technologies). For BM-derived MSCs, mononuclear cells were isolated from fresh BM aspirates (AllCells LLC, Alameda, CA) using Histopaque 1077/Accuspin tubes (Sigma-Aldrich) or from frozen human BM mononuclear cells (AllCells LLC) from both male and female donors between 19 and 24 years of age. Mononuclear cells or hemangioblasts were plated at 50,000 cells/cm2 onto tissue culture plastic in MSC growth medium as p0. Cells at p0 (BM-MSCs or hESC-MSCs) were split when large patches of adherent cells grew; cells from p1 onward were split when ∼70%–80% confluence was reached and replated at 4,000–7,000 cells/cm2. Cell surface markers were analyzed on an Accuri C6 flow cytometer (Accuri Cytometers, Inc., Ann Arbor, MI) according to standard procedures.
Adipogenic differentiation
hESC-MSCs were seeded in 12-well plates (control: 5,000 cells/well; differentiation: 20,000 cells/well) in MSC growth medium. For the control cells, media were changed every 3–4 days. For differentiation, media were replaced after 24 h with Complete Adipogenesis Differentiation Medium (STEMPRO Adipogenesis Differentiation Kit; Life Technologies), and cells were cultured for 20 days with feeding every 3–4 days. After 20 days, cells were rinsed with PBS, fixed with 4% formaldehyde for 30 min at room temperature (RT), rinsed twice with PBS, and stained with LipidTOX Green (1:100, HCS LipidTOX Neutral Lipid Stains; Life Technologies) for 30 min at RT. SlowFade Gold (SlowFade Gold Antifade Reagent; Life Technologies) was applied, and staining was visualized under a Nikon Eclipse TE2000-S fluorescent microscope (10×) using Spot Advanced software (Sterling Heights, MI).
Osteogenic differentiation
hESC-MSCs were seeded in 12-well plates (control: 5,000 cells/well; differentiation: 20,000 cells/well) in MSC growth medium. For the control cells, media were changed every 3–4 days. For differentiation, media were replaced after 24 h with Complete Osteogenesis Differentiation Medium (STEMPRO Osteogenesis Differentiation Kit; Life Technologies), and cells were cultured for 34 days with feeding every 3–4 days. After 34 days, cells were rinsed with PBS, fixed with ice-cold 70% ethanol for 1 h at RT, rinsed twice with water, stained with 500 μL 20 mg/mL Alizarin Red (Sigma-Aldrich) for 30 min at RT, and rinsed again four times with water. Calcium deposits were visualized with bright field microscopy (10×).
Chondrogenic differentiation
2.5×105 hESC-MSCs were centrifuged at 500 g for 5 min in a 15 mL conical tube, and pellets were resuspended in 0.5 mL of chondrogenic medium [high-glucose DMEM, 1 mM Sodium Pyruvate (Life Technologies), 0.1 mM ascorbic acid 2-phosphate (Sigma-Aldrich), 0.1 μM dexamethasone (Sigma-Aldrich), 1% ITS (Collaborative Biomedical Products, Bedford, MA), and 10 ng/mL TGFβ3], or MSC culture medium (for controls). Pellet cultures were maintained for 21 days with medium changes every 2–3 days, then fixed with 4% paraformaldehyde (PFA), and dehydrated in ethanol. Pellets were embedded in paraffin (Mass Histology Service, Inc., Worcester, MA), sectioned, stained with 1% Alcian Blue 8GX (Sigma-Aldrich), and counterstained with Nuclear Fast Red (Acros Organics, Morris Plains, NJ). Images were taken on an Olympus BX51 microscope with a 40×UPlanFl (NA=0.75) objective and stitched with image processing software (Fiji, open source software,
Glycosaminoglycan quantification
Sulfated glycosaminoglycan (sGAG) content was measured using a Blyscan assay (Biocolor Ltd., Carrickfergus, United Kingdom) according to the manufacturer's instructions. Briefly, 2D cultures and 3D pellets were digested with Papain (Sigma-Aldrich) before the addition of Blyscan dye reagent to precipitate the sGAG-dye complex. Samples were evaluated at 656 nm on a SpectraMax M5 plate reader against a reference standard. DNA concentration was quantified with Hoechst 33258 (Fluorescent DNA Quantitation Kit; BioRad Laboratories, Hercules, CA) using a reference standard. sGAG content was then normalized to DNA content and reported as GAG/DNA (μg/μg).
IDO activity
Cells were stimulated with IFNγ (50 ng/mL) for 3 days, lysed with NP40 lysis buffer (Life Technologies), mixed 1:1 with 2×IDO buffer (PBS with 40 mM ascorbate, 20 μM methylene blue, 200 μg/mL catalase, and 800 μM
Cytokine secretion
For most experiments, 17.7×103 cells/cm2 MSCs were plated in six-well plates for 24 h before stimulation and analyzed with cytometric bead arrays (CBA; BD Bioscience) and ELISAs (ebioscience, San Diego, CA and RayBiotech, Norcross, GA). For PGE2, MSCs were plated at 5×103 cells/cm2 in six-well plates for 24 h before stimulation. Supernatants were stored at −20°C until assays were performed according to each manufacturer's protocol.
HO-1 intracellular staining
MSCs were cultured for 3 days in Lab-Tek II Chamber Slides (Nunc). Cell fixation and permeabilization was performed with 4% PFA at RT for 10 min and 0.05% Tween-20 in 1% BSA in 1×PBS solution, respectively. Anti-human HO-1 antibody (1:100; Novus Biologicals, Littleton, CO) or IgG isotype control (1:100; BD Pharmingen) staining was performed overnight at 4°C. Cells were washed, counterstained, and mounted with DABCO/DAPI [2.5% 1,4-diazabicyclo[2.2.2]octane (Sigma-Aldrich), 50% glycerol (Fisher Scientific, Forest Lawn, NJ), and 0.005% 4′,6-diamidino-2-phenylindole (Sigma-Aldrich) in PBS)], and fluorescent images were acquired with a 20×UplanFluor objective (NA=0.45) on a Nikon Eclipse TE2000-S microscope and analyzed with Fiji open source software.
Dendritic cell maturation and MLR analyses
Dendritic cells (DCs) were derived from peripheral blood mononuclear cells (PBMCs) by a 24 h selective adherence of monocytes followed by culturing adherent cells in Lympho Medium: IMDM+10% heat-inactivated fetal calf serum containing granulocyte macrophage colony-stimulating factor (GM-CSF) and interleukin 4 (IL-4), as has been routinely used (reviewed in Refs. [53,54]), plus stem cell factor, FL, and interleukin 3 (IL-3). Flow cytometry for CD11c, CD209, HLA-ABC, HLA-DR, and CD40 confirmed that >90% of cells were DCs after 4 days of incubation in Lympho Medium. DCs were co-cultured with MSCs in Lympho Medium for 2 additional days; then, 100 ng/mL LPS+20 ng/mL IFNγ was added for 18 h to induce DC maturation (similar to Ref. [13]). Cells were subjected to flow cytometry for CD83 expression or conditioned medium (CM) was used for IL-12p70 ELISA (ebioscience) according to the manufacturer's protocol. In the “two-way” mixed leukocyte reaction (MLR), hESC-derived MSCs were used as inhibitors and DCs were used as stimulators. Non-adherent PBMCs [labeled with carboxyfluorescein succinimidyl ester (CFSE)] were used as responders. In some experiments, responders were tracked with BrdU labeling instead of CFSE. MSCs and DCs were mitotically inactivated with 5–10 μg/mL Mitomycin C (Sigma-Aldrich) before MLR co-culture. DCs were allogeneic to PBMC responders. Proliferation of responders was measured with flow cytometry for CFSE decay, CD4, and CD8 or by BrdU intracellular flow along with CD4 and CD8a, according to the manufacturer's instructions (BD Bioscience).
Animal models
For lupus nephritis (LN), 24-week-old lupus-prone (NZB×NZW) BWF1 female mice were intravenously injected with 100 μL of PBS only, 2 million hESC-MSCs in PBS, or 2 doses of 0.5×106 hESC-MSCs in PBS separated by a 2 week interval (n=10 per group). Mice received no other treatment, and survival was monitored for 70 days post-MSC injection and plotted on a Kaplan–Meier graph. Mice were from Jackson Labs (Bar Harbor, ME). For the first experimental autoimmune uveitis (EAU) experiment, 7–9-week-old C57BL/6 female mice were immunized with 500 μg of the uveitogenic peptide (amino acids 1–20) from the interphotoreceptor retinoid-binding protein (IRBP) and 1.5 μg pertussis toxin on day 0 to induce mild uveitis. hESC-MSCs (5×106/mouse) were intraperitoneally injected on day 0. On day 21, fundoscope imaging and clinical evaluation was performed in a blinded fashion by two independent clinicians. Clinical exam scores are based on a scale of 0–4, as previously described [55], with higher numbers being more severe in terms of inflammation and tissue damage. Mice were then sacrificed; eyes were harvested and fixed with 4% glutaraldehyde for 1 h and then 10% formaldehyde for 18 h; paraffin embedded; and sectioned for hematoxylin–eosin (H&E) staining. For the second EAU experiment, 7–8-week-old B10RIII female mice were immunized with 100 μg IRBP (aa161–180) peptide+1.5 μg pertussis toxin on day 0. On day 7 post-immunization, 5×106 hESC-MSCs were intraperitoneally injected per mouse and mice were sacrificed on day 14 for histology as described earlier. H&E images were captured on an Olympus BX51 microscope.
Statistical analysis
Two-tailed Student's t-tests were used to determine P-values for significance, where indicated.
Results
In order to evaluate the utility of hemangioblasts for generating MSCs, we differentiated pluripotent MA09 hESCs into EBs for 4 days followed by several days of growth in a cytokine-rich, serum-free, methylcellulose-based medium as previously described [51]. Within 3–4 days in the methylcellulose medium, clusters of shiny, spherical cells (hemangioblasts) began emerging and expanded rapidly over the course of several days, as previously described [51,52] (Fig. 1A). Within 9–10 days, hemangioblast cultures routinely expanded anywhere from 3- to 22-fold over the input EB cells (Fig. 1B). Hemangioblasts were then plated onto matrigel-coated plates in αMEM+20% FCS to enable differentiation into passage 0 MSCs. Within 5 days, patches of adherent fibroblast-like cells began emerging and represented about 15%–20% of the input cells. Adherent cells were harvested and replated for continued differentiation into passage 1 MSCs, while non-adherent cells were discarded. From p1 onward, cells were routinely passaged when cultures became ∼70%–80% confluent. A flow cytometric comparison between hemangioblasts and the resulting MSCs highlights the differences in their immunophenotype and the generation of hESC-derived MSCs with characteristic MSC cell surface markers (Fig. 1C). While 0%–10% of hemangioblasts are positive for CD90, CD105, or CD73, ≥95% of MSCS are positive for these markers. In addition, 80% of hemangioblasts are positive for the hematopoietic marker CD45, while MSCs are negative. We also tested the trilineage differentiation potential of our hESC-MSCs and found that they were able to differentiate into lipid-containing adipocytes, calcium-containing osteocytes, and GAG-containing chondrocytes (Fig. 1D–G). Thus, our hESC-MSCs met the minimal ISCT criteria to define them as MSCs [56].
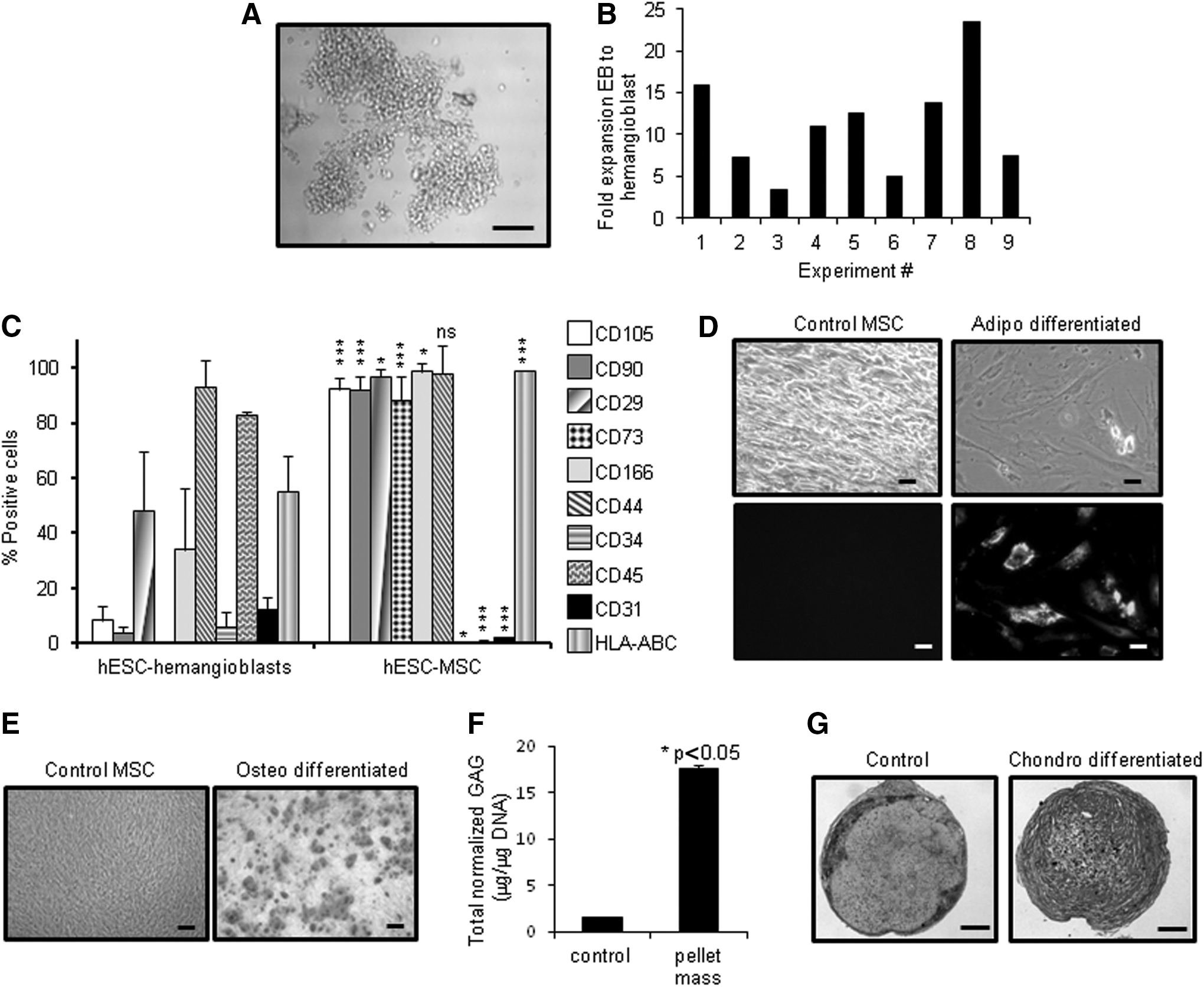
Generation of human embryonic stem cell (hESC)-mesenchymal stem cells (MSCs) using a hemangioblast intermediate step.
To further characterize our hESC-MSCs, we wanted to compare them against those derived from adult tissue. Since adult BM is arguably the most widely used source, we isolated BM-MSCs from several different donors and compared their population doublings, cell size, and cell surface markers with those of our hESC-MSCs. As shown in Figure 2A, we found that hESC-MSCs had a higher proliferation index than BM-MSCs; they underwent twice as many population doublings (for a 30,000-fold greater expansion) than BM-MSCs in just more than 2 months' time. We also noted that their size tended to be smaller than BM-MSCs at the same passage (Fig. 2B and Supplementary Fig. S2A; Supplementary Data are available online at
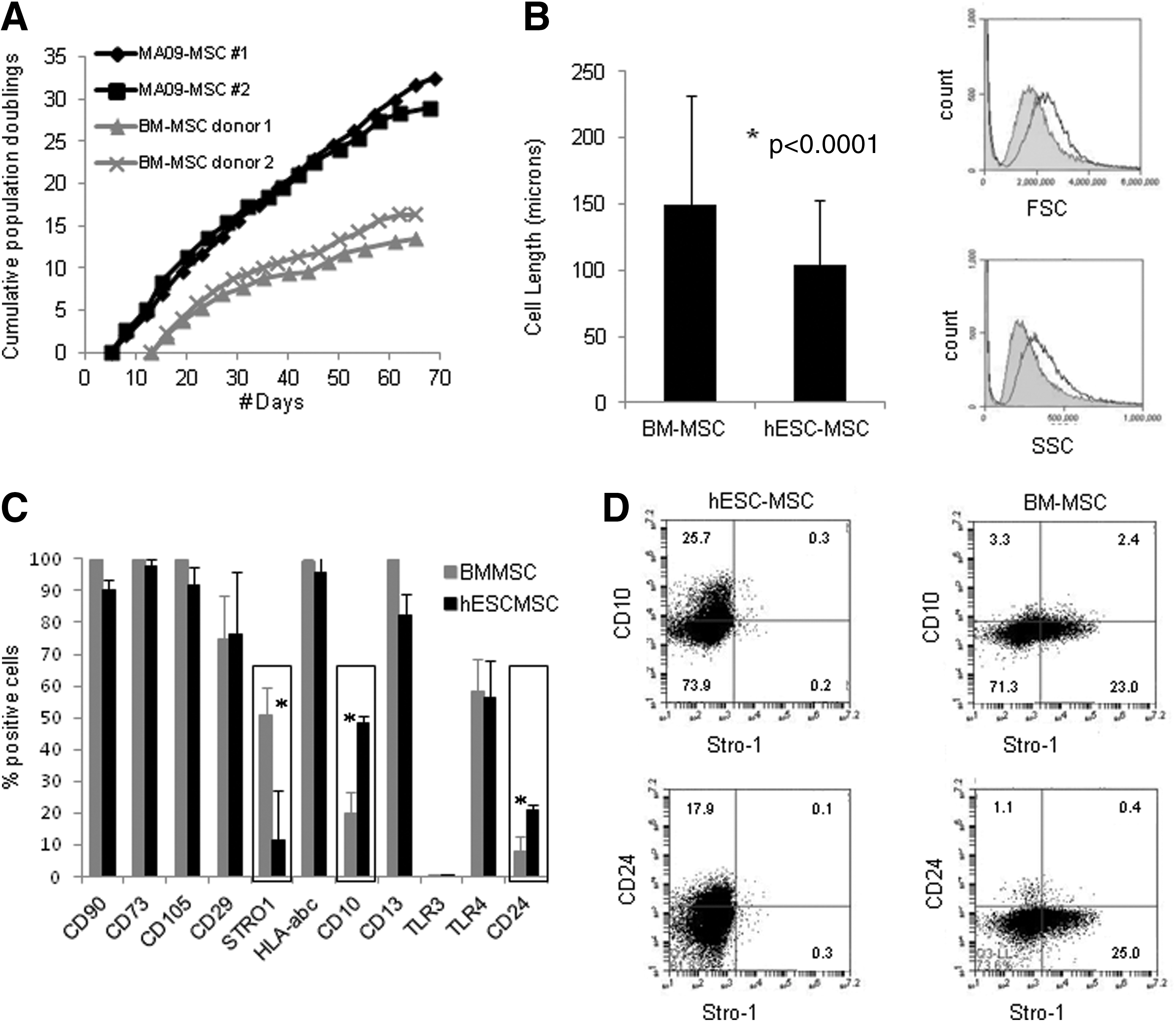
Comparison of hESC-MSCs to bone marrow (BM)-MSCs shows some similarities and some differences.
MSCs are known to secrete a panoply of cytokines and growth factors, some of which change in response to local environmental cues, and these factors are thought to alter the behavior of local immune cells and/or enhance tissue repair. We evaluated the ability of hESC-MSCs to respond to the proinflammatory cytokines IFNγ, tumor necrosis factor alpha (TNFα), or both by changes in their secreted factors. After a 48-h stimulation, we subjected MSC CM to CBA and ELISA (Fig. 3). The secreted factors, IP-10 and MIG showed some responsiveness to IFNγ stimulation but were strongly up-regulated in response to the combination of IFNγ and TNFα (first two graphs). Interleukin 6 (IL-6) was up-regulated to varying degrees in response to all three stimuli used. IL-8, MCP-1, and CCL5/RANTES (regulated on activation, normal T cell expressed and secreted) were not stimulated at all by IFNγ alone but were responsive to TNFα and the combination of TNFα and IFNγ. GM-CSF was up-regulated specifically in response to TNFα, but not when used in combination with IFNγ. Lastly, TGFβ, while detectable, was constitutively expressed and appeared to be slightly down-regulated in response to the combination of IFNγ and TNFα. Other cytokines and chemokines such as IL-4, M-CSF, IL-10, hepatocyte growth factor (HGF), VEGF, insulin-like growth factor 1 (IGF1), placenta growth factor (PIGF), bFGF, and stromal cell-derived factor 1 (SDF-1) were tested via a combination of CBA, ELISA, cytokine antibody arrays, and/or qRT-PCR but were not consistently detected or only at negligible levels (Supplementary Fig. S3A–C, and data not shown). Angiopoietin 1; brain-derived neurotrophic factor were tested by cytokine antibody arrays only, however their levels were consistently low and did not change much after stimulation (Supplementary Fig. S3A, B). We also evaluated the ability of hESC-MSCs to produce exosomes, which are microvesicles that are used to package secreted proteins, RNA, and miRNA. We followed a standard ultracentrifugation isolation procedure followed by CD63 gold immunolabeling [57] to visualize exosomes under transmission electron microscopy. Spherical vesicles of 50–75 μm in diameter were observed and positively labeled with CD63 (Supplementary Fig. S3D). These observations confirmed that the hESC-MSCs produce exosomes as a part of their secretory program.
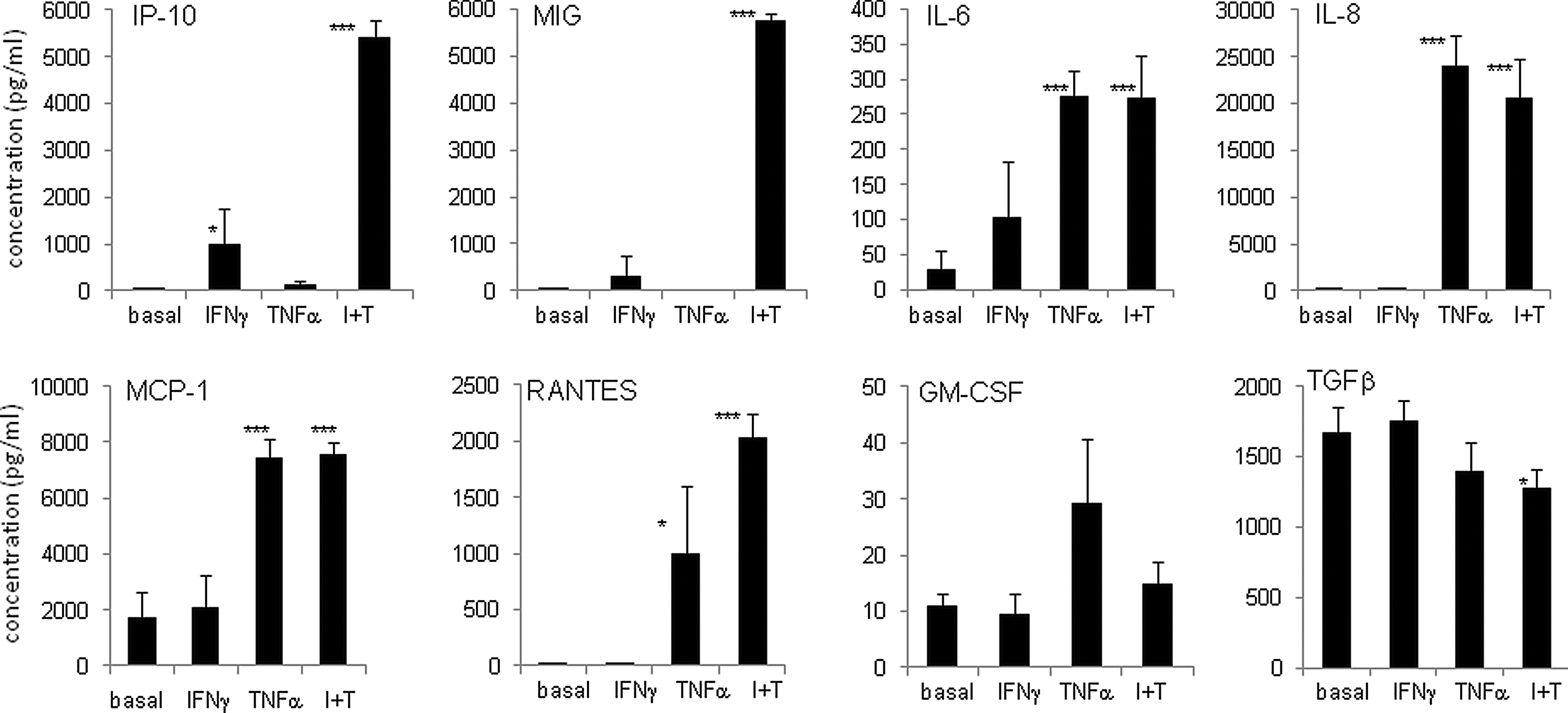
Dynamic changes in secreted proteins in response to pro-inflammatory stimulation. Quantitative assessment of cytokine concentration in hESC-MSC conditioned medium (CM) in the absence or presence of 50 ng/mL interferon gamma (IFNγ) and/or 25 ng/mL tumor necrosis factor alpha (TNFα) as indicated for 48 h. Human chemokine cytometric bead arrays (BD Bioscience) were used for inducible protein 10 (IP-10), monokine induced by gamma interferon (MIG), interleukin 8 (IL-8), monocyte chemotactic protein 1 (MCP-1), and RANTES; while ELISAs (ebioscience) were used for IL-6, granulocyte macrophage colony-stimulating factor (GM-CSF), and transforming growth factor beta (TGFβ), according to the manufacturer's protocols. Media only was used to determine background in all assays. Bars show the average of three to five independent experiments for each cytokine; error bars are SD; P-values for significance between basal and stimulated (IFNγ, TNFα, or IFNγ+TNFα) states, *P≤0.02; **P≤0.002; ***P≤0.0002.
In addition to secreting immunomodulatory cytokines, MSCs are thought to influence immune cells through the actions of HO-1, IDO, and PGE2 among other factors. Heme oxygenases are enzymes that are involved in the normal breakdown of red blood cells; they catalyze the degradation of heme into biliverdin. HO-1 is an inducible form that is involved in countering oxidative damage and inflammation [58]. We found that hESC-MSCs express HO-1 constitutively; it is readily detectable in the basal state (Fig. 4A) and does not increase in response to IFNγ or TNFα stimulation (data not shown). IDO enzymatically converts tryptophan to kynurenine, and this process is thought to deplete local tryptophan pools, thus hindering T-cell proliferation [59]. hESC-MSCs do not display IDO enzymatic activity in the basal state, yet they showed a robust induction of IDO enzymatic activity in response to IFNγ stimulation, (Fig. 4B). We observed that IDO gene expression is also strongly up-regulated in response to co-culture with mitogen-activated PBMCs (Supplementary Fig. S3E). PGE2 is an eicosanoid molecule that is produced through multi-step metabolism of arachidonic acid and is thought to play a role in the anti-inflammatory and immunomodulatory effects of MSCs, including the inhibition of DC maturation [8]. We found that PGE2 secretion from hESC-MSCs is strongly up-regulated in response to costimulation with IFNγ and TNFα. Collectively, these data demonstrate that our hESC-MSCs produce a variety of bioactive proteins and they can respond to local environmental cues in a manner which is consistent with what has been reported for adult tissue-derived MSCs [60].
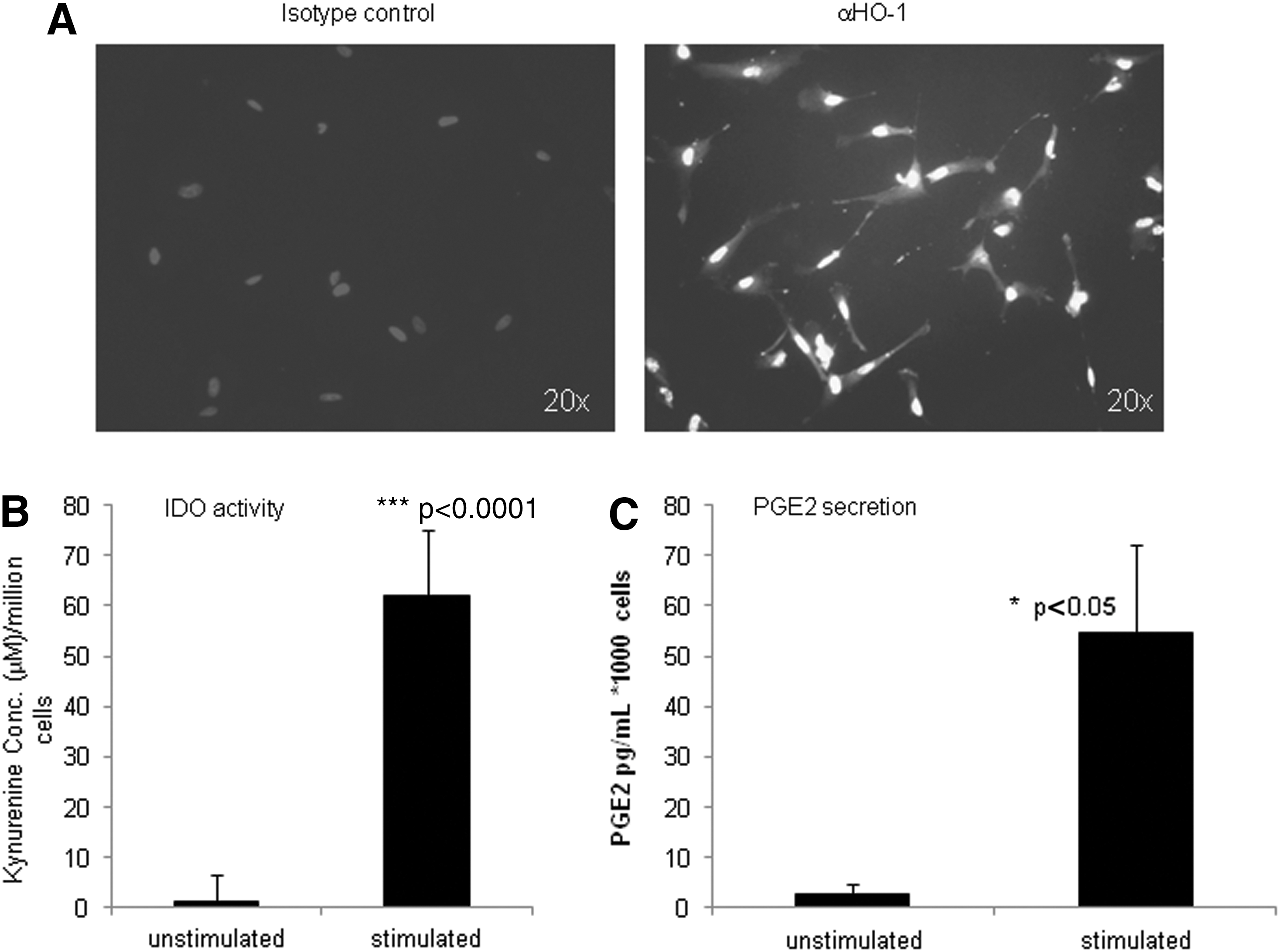
Signaling molecules produced by hESC-MSCs.
hESC-MSCs, equipped with an immunomodulatory arsenal, should be able to affect the activity of both innate and adaptive immune cells directly. To test this, we generated immature DCs from PBMC monocytes using a medium containing GM-CSF and IL-4. Flow cytometry confirmed expression of CD11c, CD209, HLA-DR, and CD40 on ≥95% of cells after 4 days of differentiation (Supplementary Fig. S4A–C). CD11c versus CD73 staining could also be used to distinguish DCs from MSCs in co-culture (Supplementary Fig. S4C, D). In the absence of MSCs, DCs strongly up-regulated both CD83 expression (Fig. 5A) and IL-12p70 secretion (Fig. 5B) in response to a LPS/IFNγ maturation cocktail; however, co-culture with MSCs caused a reduction in the inducible CD83 and IL-12p70 expression on exposure to maturation cocktail. We next tested the ability of hESC-MSCs to interfere with T-cell proliferation in response to different stimuli by using MLRs assays. Non-adherent PBMCs were used as responders, allogeneic immature DCs were used as stimulators, and MSCs were used as third-party effectors. In response to increasing amounts of DCs, the CD4+ and/or CD8+ T-cell population within the PBMCs proliferated in a DC dose-dependent manner, as assessed by BrdU staining (Fig. 5C, gray line with squares). When a fixed amount of MSCs were added to the MLR, they inhibited the DC-induced T-cell proliferation (Fig. 5C, black line with diamonds and Supplementary Fig. S5). When a constant amount of allogeneic DC was used as stimulation, increasing amounts of MSCs inhibited T-cell proliferation in a dose-dependent manner (Fig. 5D and Supplementary Fig. S6). Likewise, when PMA and ionomycin were used as a mitogenic stimulus, increasing amounts of MSCs also inhibited T-cell proliferation in a dose-dependent manner (Supplementary Fig. S7). MSCs were also observed to enhance the CD4+/CD25+/FoxP3+ regulatory T-cell population in response to IL-2 stimulation of PBMCs (Supplementary Fig. S8). These data show that hESC-MSCs are able to impact both innate and adaptive immune cells in response to different stimuli.
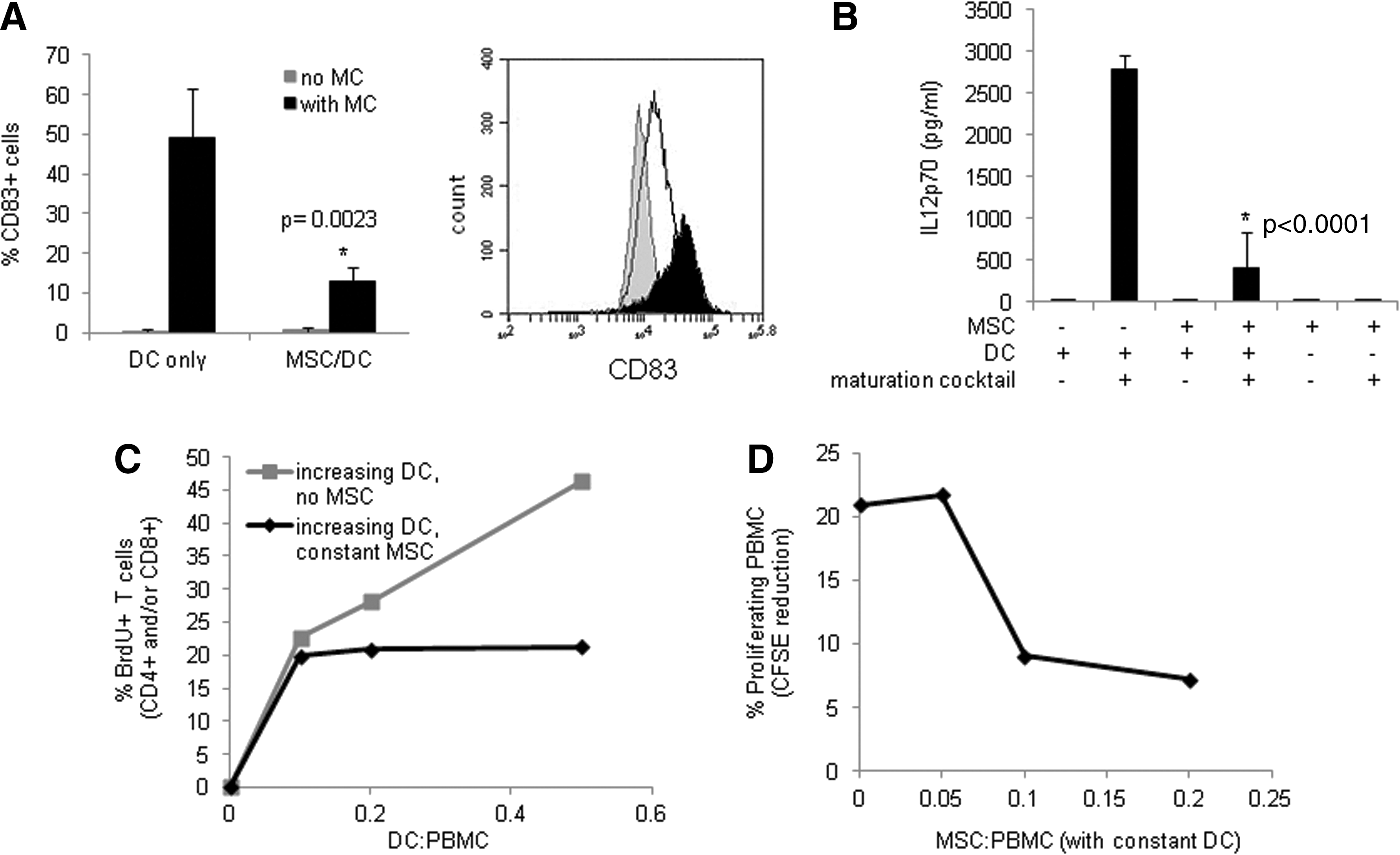
Immunomodulatory activity of hESC-MSCs on cells of innate and adaptive immune system.
The true litmus test for MSC clinical potential is an in vivo demonstration of efficacy in combating disease. We used two different autoimmune disease mouse models to examine hESC-MSC therapeutic utility: LN and EAU. LN spontaneously arises in BWF1 mice (F1 offspring from NZW×NZB cross), beginning at around 20–24 weeks of age [61] and resulting in a reduced lifespan (50% mortality by ∼35 weeks) due to kidney dysfunction [62] (Fig. 6A, black line with diamonds). We found that the administration of hESC-MSCs, as either a single injection (Fig. 6A, black line with squares) or two injections given 2 weeks apart (Fig. 6A, gray line with triangles), significantly prolonged the lifespan of BWF1 mice. Further investigation into the mechanism(s) underlying MSC therapeutic effects is an active area of investigation. EAU is an inflammatory eye disorder of the uvea, which can be induced in mice by immunizing with peptides from the retinal protein, IRBP [63]. We induced mild uveitis in C57BL/6 mice and assessed disease severity in a blinded fashion using both in vivo fundoscopic imaging and histopathology. Both of these examinations are scored on standard scales of 0–4, with the higher number indicating more severe phenotype [55]. EAU mice that received hESC-MSCs had significantly lower disease scores than EAU mice which did not receive hESC-MSCs (Fig. 6B, C). In addition, we injected MSCs into C57BL/6 mice that were not induced with EAU and found no evidence of histologic changes in eyes, brain, lungs, spleen, and GI tract (data not shown). In a pilot study of more severe EAU performed in B10RIII mice, a single injection of hESC-MSCs markedly reduced disease severity as well. Histologic eye images of B10RIII EAU mice that did not receive any hESC-MSCs show heavy inflammatory cell infiltrates in the vitreous cavity, multiple retinal folds and detachment, inflammatory cell infiltration with disruption of the outer retina, and subretinal neovascularization (Fig. 6D left panel). In comparison, EAU mice treated with hESC-MSCs (Fig. 6E, right panel) show far fewer inflammatory cell infiltrates in the vitreous space, reduced retinal folds, and less tissue damage. A more detailed study is underway to probe the mechanisms underlying the ability of hESC-MSCs to reduce clinical features of EAU. Together, the LN and EAU data provide proof of principle that our hESC-MSCs have therapeutic utility against autoimmune disorders.
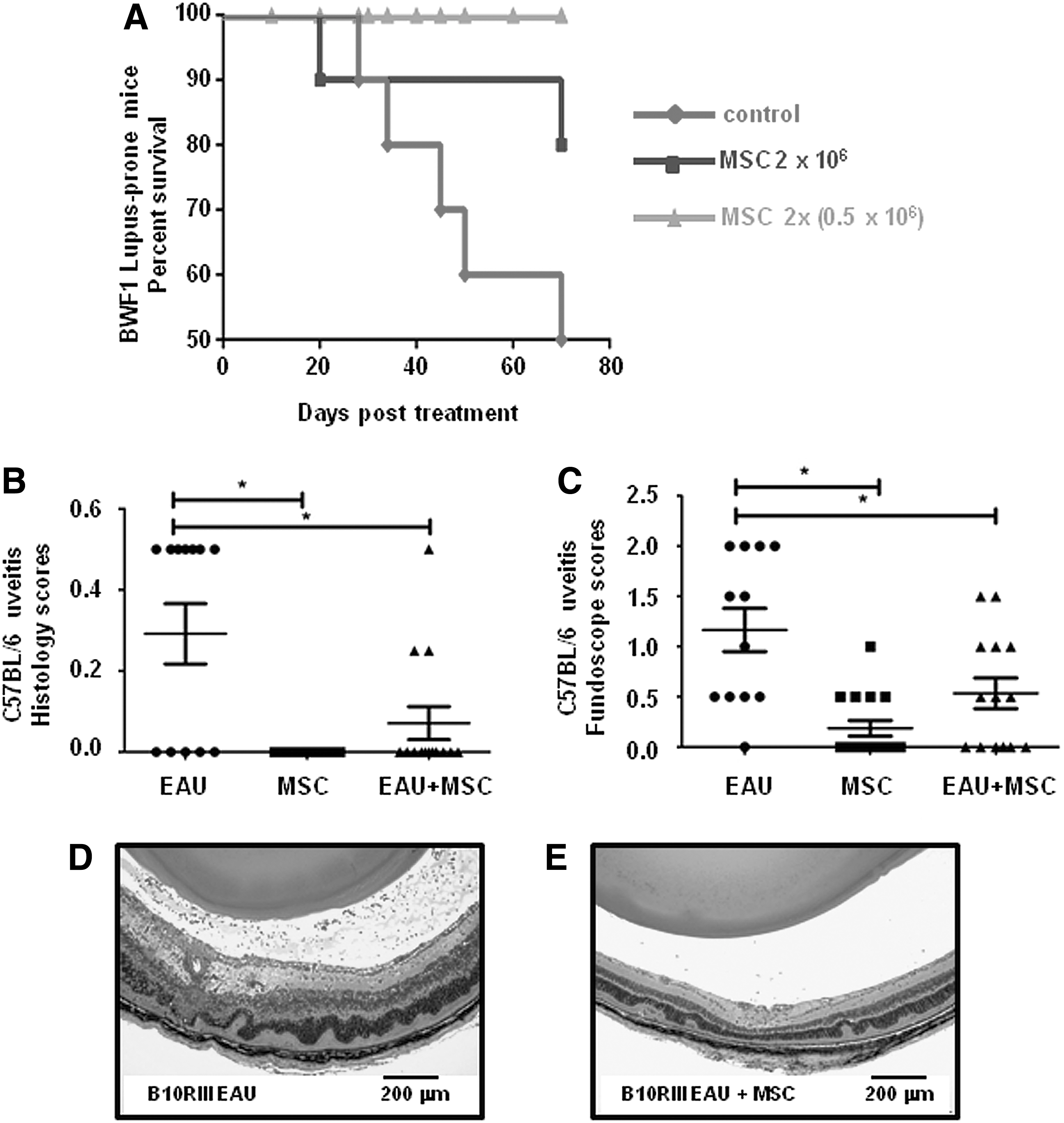
Proof of principle in vivo testing for hESC-MSC therapeutic activity in two autoimmune disease mouse models.
Discussion
Several different methods have been used to generate MSCs from PSCs, yet their immunomodulatory properties and in vivo therapeutic potential have not been nearly as well characterized as those of BM-MSCs. We describe here the use of hemangioblasts as an intermediate cell type to bridge the differentiation process between hESCs and MSCs. While extremely transient during development, hemangioblasts can be generated [51,64,65] and easily expanded from hESCs in vitro [52]. Their potential as an endothelial/hematopoietic precursor has proved to be quite useful for generating vascular endothelium and smooth muscle [51,66,67], red blood cells [68], platelets [69], DCs (our own unpublished data), and now MSCs. The use of a hemangioblast intermediate helps push the initial differentiation of hESCs in a more directed fashion than the stochastic differentiation involved in exclusively using EBs. It also provides an opportunity to expand the pool of progenitors that may give rise to MSCs before further differentiation, a property which is important when one considers that not all hESCs will turn into MSCs. Moreover, going through a hemangioblast intermediate also helps avoid possible carry-over of residual pluripotent cells, a risk that direct plating methods cannot easily avoid but which is an important consideration for any cellular therapy derived from hESCs.
In surveying the properties of our hESC-MSCs that may give them therapeutic potential, we found that they produce a dynamic array of cytokines, chemokines, and growth factors, including IL-10, CCL2, and GM-CSF, MIG, IP-10, HO-1, IDO, and PGE2, many of which change depending on signals in the environment. These factors are overlapping with those secreted by adult tissue-derived MSCs [6,8,70], cord-blood-derived MSCs [71], and other hESC-MSCs [72]. It is likely that the combination of many secreted factors contribute to MSC therapeutic effects in different in vivo environments. Adult tissue-derived MSCs have been shown to inhibit DC differentiation and maturation [13,14], yet this study is the first to examine the effects of hESC-derived MSCs on DCs. Our hESC-MSCs suppressed CD83 expression and IL-12p70 secretion, both of which are involved in the DC maturation/activation process and integral to the ability of DCs to properly deliver signals to T cells [73]. This is also the first study which shows that hESC-MSCs can enhance regulatory T-cell populations in response to IL-2, similar to what has been reported for BM-MSCs [17]. Three other groups which have generated hESC-MSCs reported that their cells inhibited T-cell proliferation in MLR assays [38,41,44] as did ours in response to both allogeneic DCs and mitogenic stimuli. However, a fourth group found that their hESC-MSCs did not inhibit T-cell proliferation in MLR assays [48]. Obviously, differences in the starting hESC lines and MSC derivation methods may affect the quality and properties of the resulting cells. It may be interesting to do a side-by-side comparison of immunomodulatory properties for hESC-MSCs derived with different methods. Nonetheless, our hESC-MSCs displayed a range of features and functions similar to those reported for BM-MSCs and considered important for MSC medicinal effects.
Despite many similarities to BM-MSCs, we identified some differences as well. We observed that approximately 25% of BM-MSCs expressed Stro-1, yet few, if any, hESC-MSCs expressed it. One study suggests that Stro-1 expression is indicative of the starting tissue, with it being expressed in MSCs derived from BM but not in those derived from adipose [74]. However, another study finds that Stro-1 is important for a high proliferative rate of mesenchymal progenitors from BM [75]. We found a high proliferative rate of our MSCs without Stro-1 expression, thus underscoring the possibility that other markers or properties, perhaps particular to individual tissue sources, can have similar functional consequences as Stro-1 may have for BM. We found that both CD10 and CD24 expression was greater in hESC-MSC populations than in BM-MSCs. Interestingly, when cells were grown in commercially available, serum-free media instead of standard aMEM+20% FCS, CD10 became highly expressed (≥90%) in both MSC types (data not shown), indicating that the expression of certain cell surface markers may be subject to changes in media components. For CD24, we noted that its expression at both the RNA and protein level declined with age or on exposure to IFNγ (data not shown). CD24 is an alternative ligand for p-selectin, is mutated in several autoimmune disorders, and has pleiotropic roles depending on the cell type in which it is expressed [76]. Lian et al. used CD24 as a negative marker for sorting MSC progenitors (CD105+, CD24−) from hESC cultures [40]. The observation that a subset of our hESC-MSCs maintains its expression for several passages under basal conditions is intriguing, although its significance is not entirely clear. Preliminary sorting experiments show that CD24+ and CD24− hESC-MSCs proliferate at roughly the same rate (data not shown), yet testing the potential difference in their in vivo functionality may provide some indications as to its biological relevance. We also observed that the proliferative capacity of our hESC-MSCs is far greater than that of BM-MSCs, which is similar to what has been reported for other hESC-MSCs [41].
Previously, BM-MSCs have been observed to increase in size as they age [77]; we also observed that hESC-MSCs got larger with successive passaging, yet they tended to be smaller than passage-matched BM-MSCs, even at later passages. MSC function (differentiation potential, migration, and antioxidant capacity among others) is thought to decline as their growth slows down and they senesce [78,79]. A greater proliferative capacity and smaller size may indicate that hESC-MSC functionality can be maintained longer, enabling more therapeutic cells to be derived from a single lot of starting material than BM-MSCs.
Lastly, and unlike adult tissue-derived MSCs, a few studies have explored the therapeutic utility of hESC-MSCs in disease models. TNBS-induced experimental colitis in mice was the first model used to show that hESC-MSCs have an in vivo therapeutic function comparable to adipose- and cord-blood-derived MSCs [41]. hESC-MSCs have also been found to home to ischemic limbs [42] and enhance hematopoietic stem cell engraftment [48] in mouse models. Here, we examined the potential clinical effects of hESC-MSCs in two different autoimmune disorder models, LN and EAU.
For LN, adipose- [80] and cord-blood-derived MSCs [81] have been shown to have therapeutic effects in lupus-prone BWF1 mice while another report indicates that BM-MSCs do not [82]. The MSCs that worked in this model were found to delay onset of proteinuria [80,81], increase Treg populations [80], decrease anti-dsDNA [81], and affect the balance of Th1/Th2 cells [81], leading to preservation of kidney function and an increase in lifespan [80,81]. Our cells similarly afforded a great increase in average lifespan, and investigations are underway to probe the molecular mechanisms behind this clinical improvement. Similarly, in pilot studies for both mild and severe EAU, our hESC-MSCs were found to decrease the severity of disease as determined by fundoscopic imaging and histologic examination. Previous studies using murine BM-MSCs indicate that alterations in cytokine secretion and the balance between pro- and anti-inflammatory T-cell subsets (eg, Th1/Th2, and Th17/Treg) may play a role in MSC therapeutic activity in EAU models [83 –85]. A more detailed study is currently being performed to confirm these initial observations and should shed light on the mechanisms by which our hESC-MSCs may achieve clinical improvement. While research indicates that different sources of MSCs may have different properties [86,87], the data collectively presented here illustrate that hESC-MSCs are a viable alternative to adult-derived MSCs with the added benefit that they can circumvent many problems associated with adult-tissue derived MSCs. Additional studies in other animal disease models can help define the range of indications that hESC-MSCs may be useful in treating and may 1 day provide enough impetus to enable hESCs to rival adult BM as the preferred starting material for clinical development of MSCs.
Footnotes
Acknowledgment
The authors thank Lucy Vilner for cell culture assistance.
Author Disclosure Statement
E.A.K., N.A.K., G.Y., J.C., and R.L. are employees of Advanced Cell Technology, a biotechnology company engaged in stem cell and regenerative medicine research.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.