
Abstract
To be clinically relevant as a therapy for heart failure, endogenous progenitor cells must be isolated and expanded from aged and/or diseased tissue. Here, we investigated the effect of age and cardiac impairment resulting from lack of dystrophin on murine cardiosphere-derived cells (CDCs). CDCs were isolated and expanded from atrial biopsies from wild-type mice aged 1.5, 6, 18, and 24 months and from mdx mice aged 6 and 18 months. Cardiac function was measured in mdx mice and age-matched wild-type mice using high-resolution cine magnetic resonance imaging. CDCs could be isolated and expanded from all mice, however, the number of cells obtained, and their regenerative potential, decreased with age, as demonstrated by decreased expression of stem cell markers, c-kit and Sca-1, and decreased cell proliferation, migration, clonogenicity, and differentiation. Six-month-old mdx mice showed right ventricular (RV) dilation and reduced RV ejection fraction (EF) in comparison to wild-type mice. Older mdx mice displayed significant RV and left ventricular dilation and decreased EF in both ventricles, compared with age-matched wild-type mice. Mdx mouse hearts contained significantly more fibrotic tissue than age-matched wild-type mouse hearts. However, CDCs isolated from mice aged 6 and 18 months had the same number and regenerative potential from mdx mice and age-matched wild-type mice. Thus, the cardiac progenitor cell population is impaired by age but is not substantially altered by the progressive deterioration in function of the dystrophic heart.
Introduction
D
Here, we have investigated the effect of aging and mild cardiac impairment on murine CDCs.
Methods
The study was approved by The University of Oxford Animal Ethics Review Committees and the Home Office (London, United Kingdom) under Project License 30/2278. Animals were kept under controlled conditions for temperature, humidity, and light, with water and rodent chow available ad libitum. At the end of each experiment, animals were anesthetised with sodium pentobarbital (200 mg/kg body weight, IP; Euthatal) to allow tissue removal.
Cell isolation and expansion
Mouse hearts were washed twice in phosphate-buffered saline (PBS) and the atria were cut into 5 mm segments and digested in 0.05% trypsin (Invitrogen) for 3 min at room temperature. The segments were further minced into 1 mm fragments that were washed again in PBS and plated out as explants onto fibronectin-coated (Sigma) 60 mm Petri dishes (Corning) containing 1.5 mL of Complete Explant Medium (CEM, see Supplementary Data; Supplementary Data are available online at
AlamarBlue® cell proliferation assay
The viability and proliferation of CDCs were assessed using AlamarBlue (AbD Serotec) according to manufacturer's instructions. Briefly, CDCs at passage 2 were seeded into a 96-well plate at a density of 500 cells/well and allowed to attach overnight. The cells were washed twice with PBS and incubated with 10% AlamarBlue, diluted in phenol red-free Iscove's modified Dulbecco's medium (IMDM; Invitrogen), at 37°C. The relative fluorescence intensity at 530–560 nm excitation wavelength and 590 nm emission wavelength, quantified by FLUOstar OPTIMA (BMG Labtech), was directly related to the number of live cells using a standard curve.
Clonogenic assay
CDCs at passage 2 were harvested using trypsinization to produce a dissociated single-cell suspension that was serially diluted to 50 cells in 10 mL of CEM and seeded into a 96-well plate at a density of 0.5 cell per well to generate single-cell clones. After 4 h, each well containing a single cell was identified under a light microscope and examined for growing colonies twice weekly. After 2 weeks, the number of wells with clones derived from a single cell was counted. Clonogenicity of CDCs was determined using the following formula:
Clonal efficiency (%)=(Total wells with clone/Total wells with single cell)×100.
Flow cytometry
Once confluent at passage 2, CDCs were harvested using trypsin (5 min at 37°C), washed with PBS, and fixed with 4% paraformaldheyde (Sigma) for 10 min. For intracellular markers the cells were treated with 0.2% Triton X-100 in PBS for 10 min. The samples were incubated with 2% bovine serum albumin (BSA)+2% fetal bovine serum (FBS) in PBS for 30 min and then treated with primary antibodies for 1 h (see Supplementary Table S1 for antibodies used). After rinsing with PBS, cells were treated with the secondary antibody for 30 min. The samples were kept at 4°C in the dark prior to analysis using a FACSCalibur flow cytometer (BD Biosciences). The data were analyzed using Summit4.3 software.
Telomere length and telomerase expression
Relative telomere lengths were determined using quantitative real-time polymerase chain reaction, as described previously [28]. Total DNA was extracted from cultured cells using the QIAGEN® Blood and Cell Culture DNA kit (Qiagen GmbH) according to the manufacturer's instructions. To evaluate the purity and concentration of DNA, absorbances were measured at 260 nm (A260) and 280 nm (A280) using a Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies, Inc.). Telomeric DNA was amplified using specific primer pairs (see Supplementary Table S2). The amplification of telomeric DNA was normalized to a single copy gene 36B4 (see Supplementary Table S2). For the comparison of telomerase expression, total cellular RNA was isolated and the telomerase expression of CDCs was determined based on the mRNA level of telomere reverse transcriptase (TERT) enzyme and normalized to GAPDH (see Supplementary Table S2 for primers).
Cardiomyogenic differentiation
Cardiomyogenic differentiation was induced using cardiomyocyte differentiation medium (CDM) [2% FBS (Invitrogen), 1% insulin–transferrin–selenium in IMDM:DMEM/F12 (1:1; Sigma)] supplemented with 1 μM 5-azacytidine. The supplemented CDM was changed every 2 days for 6 days. Then, all cells were aspirated with PBS to remove the dead cells and CDM supplemented with 1 μM ascorbic acid was added to the plate. The medium was changed every 2 days for the following 6 days.
Immunocytochemistry
Conditioned CDCs were cultured on fibronectin-coated glass slides and fixed with 4% paraformaldehyde (Sigma) for 10 min. For intracellular markers, the cells were treated with 0.2% Triton X-100 in PBS for 10 min, blocked with 2% BSA+2% FBS in PBS, and incubated with primary antibodies for 1 h (see Supplementary Table S1). After rinsing with PBS, cells were treated with the secondary antibody for 30 min. The slides were kept at 4°C and in the dark until analyzed using an Olympus FV1000 Fluoview confocal microscope.
Tissue sectioning and histology
For histology and immunohistochemistry, explanted hearts were cut into half transversely, embedded and frozen in optimal cutting temperature compound (OCT; Sakura Tissue-Tek) on dry ice, and stored at −80°C. Frozen heart tissues were sliced into 10 μm sections in thickness at −20°C using a cryostat (Bright 5040) and stored at −80°C until use. Sections were stained with picro-sirius red solution. The analysis of cardiac fibrosis was performed on five regions from each heart, including right ventricle, interventricular septum, and left ventricle (anterior wall, lateral wall, and posterior wall). Images were captured at 10 and 25× magnification on a Nikon light microscope attached to a digital Canon camera (EOS 1000D; Canon, Inc.). The percentage of cardiac fibrotic area for wild-type and mdx mice was measured using ImageJ.
Cardiac cine-magnetic resonance imaging
Cardiac cine-magnetic resonance imaging (MRI) was performed as described [29,30]. Briefly, mice were anesthetized with 1.5% isoflurane in O2 and positioned supine in a purpose-built cradle. ECG electrodes were inserted into the forepaws and a respiration loop was taped across the chest. The cradle was lowered into a vertical-bore, 11.7 T MR system (Magnex Scientific) with a 40 mm birdcage coil (Rapid Biomedical) and a Bruker console running Paravision 2.1.1 (Bruker Medical). A stack of contiguous 1 mm thick true short-axis ECG and respiration-gated cine-FLASH images (TE/TR 1.43/4.6 ms; 17.5° pulse; field of view 25.6×25.6 mm; matrix size 256×256; voxel size 100×100×1,000 μm; 20–30 frames per cardiac cycle) were acquired to cover the entire left and right ventricles.
MRI data analysis
Image analysis was performed using ImageJ (NIH Image). Left ventricular mass, volumes, and EF were calculated as described [31]. RV volumes were calculated from the same stack of short-axis cine images, but two to three extra slices at the base of the heart were required to cover the entire RV, as described by Wiesmann et al. [32].
Data and statistical analysis
Results are shown as mean±standard error. Differences were investigated using a 1 way ANOVA with a Tukey post hoc test and considered significant at P<0.05 (PASW Statistics Version 18).
Results
The effect of age on CDCs
Explants of atrial tissue from C57 black 10 mice aged 1.5, 6, 18, and 24 months (n=4 for each group) were plated on fibronectin-coated dishes. EDCs were generated from all explants, however, EDC growth decreased with increasing age. There was a significant decrease in the number of EDCs generated from 6- to 24-month-old mice by 26%, 45% and 58%, respectively, compared with 1.5-month-old mice (Fig. 1A). Further, the maximal distance EDCs migrated from the explants decreased with donor age (Fig. 1B). It was possible to form cardiospheres from EDCs of all age groups, although older mice gave significantly fewer cardiospheres (90±7, 70±6, 47±5, 43±5 cardiospheres/well were obtained from 1.5-, 6-, 18-, and 24-month-old mice, respectively; P<0.05 vs. 1.5 months old). Cardiospheres were expanded as CDCs on fibronectin to passage 2. There was a significant reduction in the number of CDCs generated from 6-, 18-, and 24-month-old mice by 25%, 44%, and 55%, respectively, compared with 1.5-month-old mice (Fig. 1C). Further, the proliferation rate of CDCs (measured using Alamar Blue) was reduced in older mice such that the number of CDCs obtained after 7 days in culture was ∼1.5, 1.8, and 2.3-fold higher from 1.5-month-old mice than from mice aged 6, 18, and 24 months, respectively (Fig. 1D).
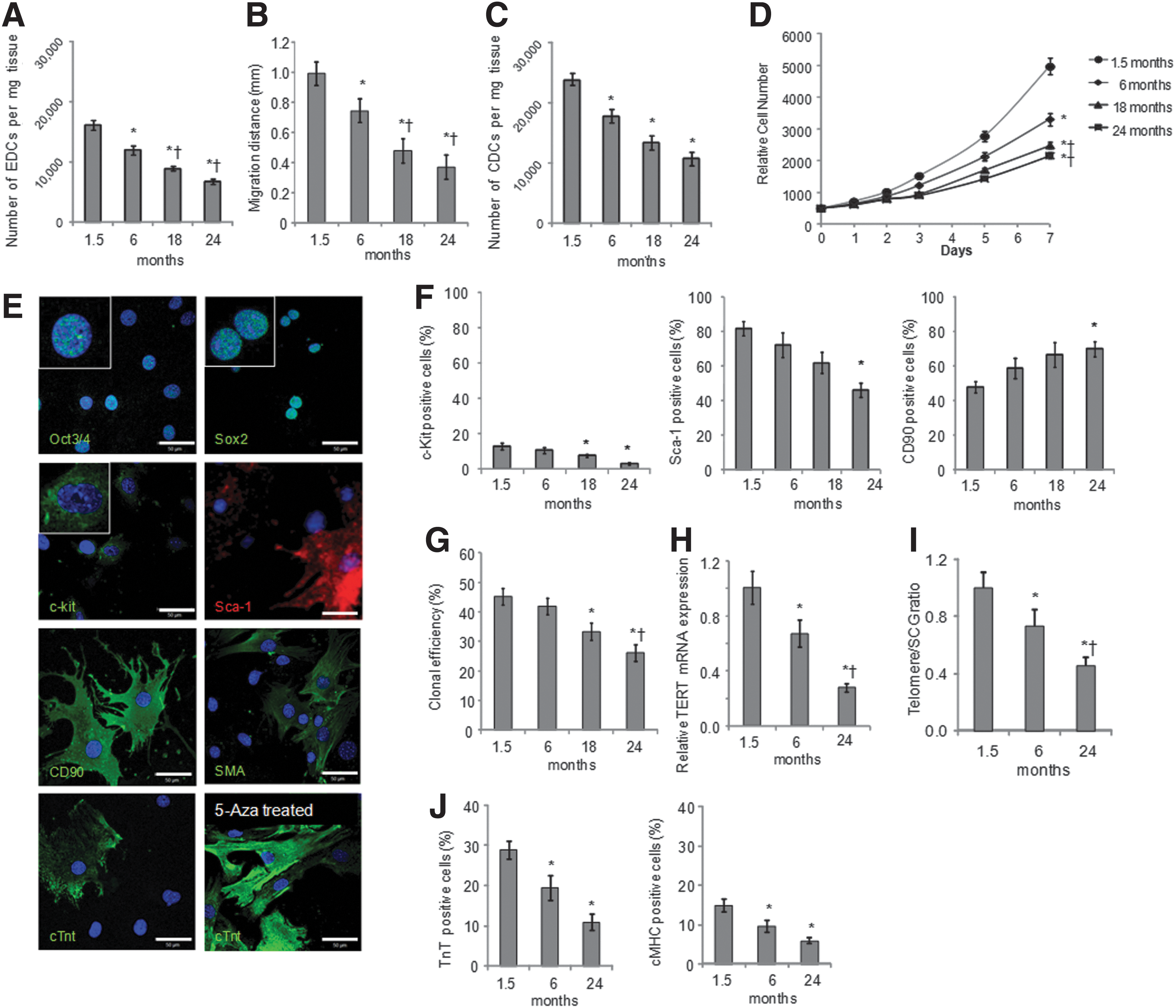
Cardiosphere-derived cell (CDC) culture from mice aged 1.5 to 24 months. Explant-derived cells (EDCs) were cultured from atria of mice aged 1.5, 6, 18, and 24 months (n=4 for each group). The number of EDCs
CDC characterization
CDCs from young mice, characterized using confocal microscopy, expressed the pluripotent markers Oct3/4 and Sox2, the CSC markers c-kit and Sca-1, and the mesenchymal marker CD90 (Fig. 1E). Some cells differentiated spontaneously, as shown by expression of smooth muscle actin and cardiac troponin T (cTnT; Fig. 1E). More robust expression of cTnT was observed after 2 weeks of treatment with 5-azacytidine and ascorbic acid (Fig. 1E). In CDCs from mice aged 1.5 to 24 months, surface marker expression of c-kit, Sca-1, and CD90 was measured using flow cytometry. Expression of the CPC markers, c-kit, and Sca-1, decreased with age such that expression was significantly lower in CDCs from 24-month-old mice than in cells from young mice (Fig. 1F). Expression of the mesenchymal marker CD90 was significantly higher in cells from very old mice than in those from 1.5-month-old mice (Fig. 1F). The clonal efficiency of CDCs also decreased with age, as CDCs from mice aged 18 and 24 months included significantly fewer cells capable of forming clones than those from mice aged 1.5 months (Fig. 1G). Further, telomere length and mRNA expression of TERT were significantly decreased in CDCs from older mice compared with those from 1.5-month-old mice (Fig. 1H, I). CDCs treated with 5-azacytine and ascorbic acid to induce cardiomyogenic differentiation showed increased expression of myocyte-specific proteins cTnT and cardiac myosin heavy chain, cMHC, confirmed by flow cytometry. Treated CDCs from mice aged 6 or 24 months contained significantly fewer differentiated cells than those from 1.5-month-old mice (Fig. 1J).
Cardiac function and fibrosis in mdx mice
In vivo cardiac function in mdx mice and age-matched C57 Black 10 wild-type controls was measured using high resolution cine MRI (Fig. 2A, B and Supplementary Table S3). At 6 months, mdx mouse hearts had RV dysfunction with an increased end systolic volume and a reduced EF. The cardiac output of the RV was reduced, although not significantly, but with a corresponding decrease in LV cardiac output that resulted from non-significant decreases in stroke volume and heart rate. At 18 months, mdx hearts showed both LV and RV dysfunction with dilation and reduced EF of both chambers. The mdx mouse hearts were hypertrophied and the resulting (non-significant) increase in stroke volume resulted in a cardiac output that was not significantly different from that of the wild-type mice.
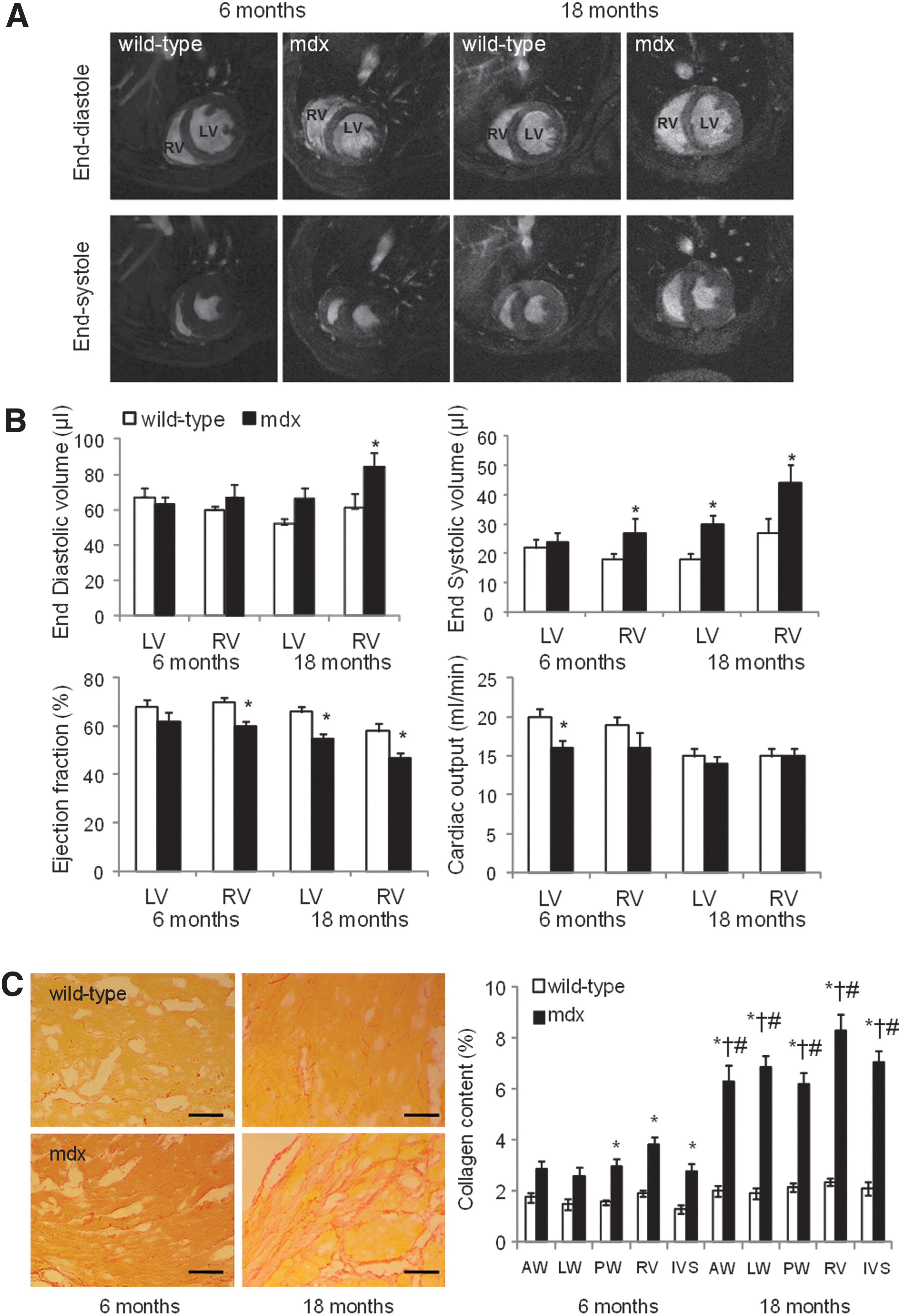
Mdx mice have impaired cardiac function. In vivo magnetic resonance imaging of hearts from wild-type and mdx mice aged 6 or 18 months
Ex vivo histology of hearts at 6 and 18 months showed that mdx mice hearts were more fibrotic than those of wild-type mice, with a 1.8-fold and 3.3-fold increase in collagen content compared with wild-type hearts, respectively, measured using picro-sirius red staining (Fig. 2C).
CDC culture from wild-type and mdx mice
Explants of atrial tissue from wild-type and mdx mice aged 6 and 18 months were plated out on fibronectin (Fig. 3A). As seen previously, significantly fewer EDCs and CDCs were obtained from 18-month-old mice than from 6-month-old mice (Fig. 3B, D). However, the number of EDCs or CDCs obtained from mdx mice were not significantly different from those obtained from wild-type mice of the same age. Similarly, the EDC migration distance and the CDC clonal efficiency were significantly lower in cells from 18-month-old mice than in cells from 6-month-old mice but there was no significant difference between cells from mdx mice and those from age-matched wild-type mice (Fig. 3C, E). CDC proliferation over 7 days was significantly lower in cells from older mice than in those from younger mice, but again there was no significant difference between the proliferation rate of CDCs from mdx mice and age-matched wild-type mice (Fig. 3F).
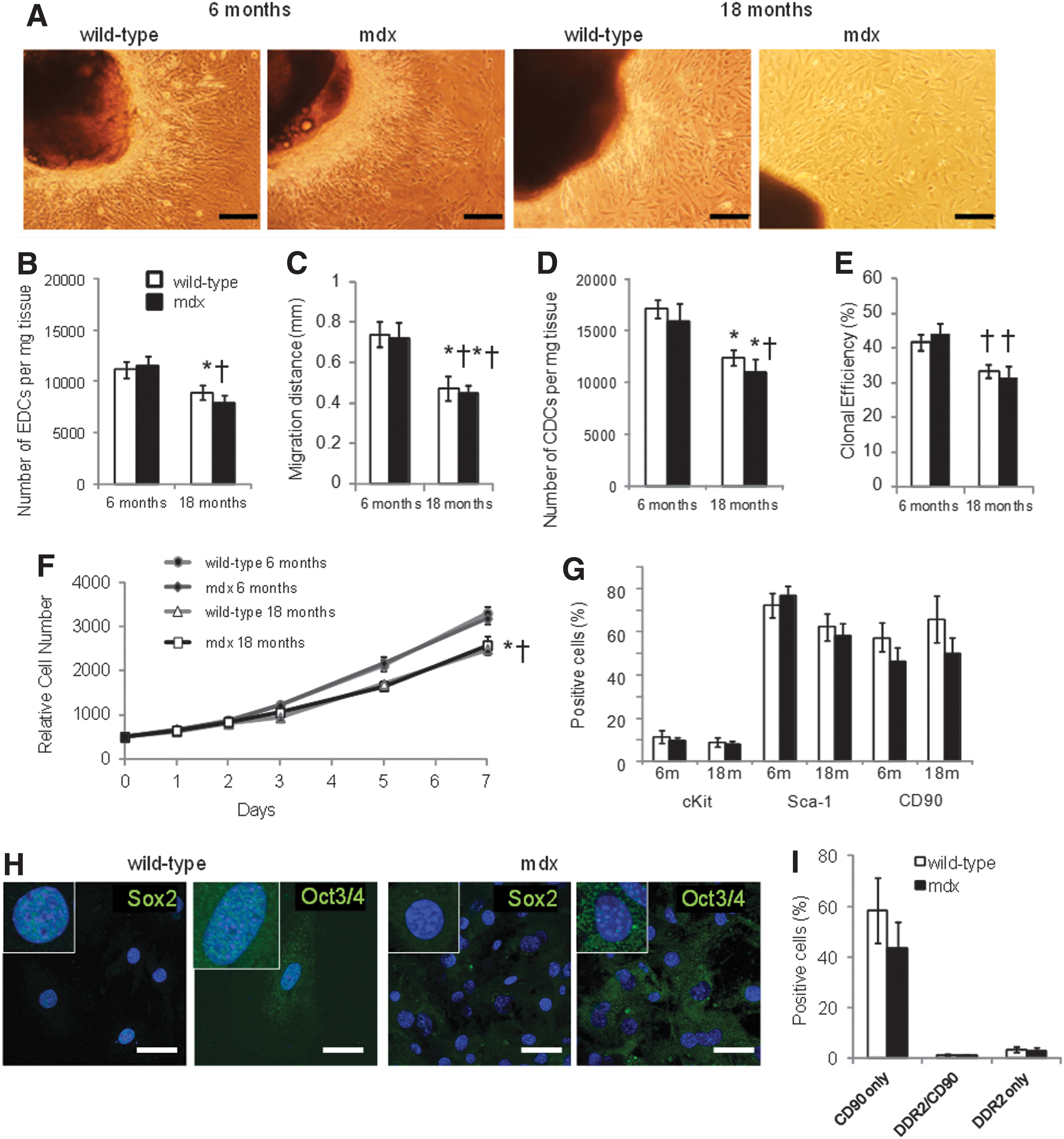
CDCs from mdx mouse hearts. EDCs and CDCs were cultured from wild-type and mdx mice aged 6 or 18 months (n=7 for each group at 6 months, n=8 for each group at 18 months), representative images of EDCs are shown in
Analysis using flow cytometry showed that there were no differences in expression of the surface markers c-kit, Sca-1, and CD90 between CDCs from wild-type and mdx mice at 6 or 18 months of age (Fig. 3G). However, nuclear expression of the pluripotent markers Sox2 and Oct3/4 was not detected in CDCs from the older mdx mice, although it could still be detected in CDCs from age-matched wild-type mice (Fig. 3H). Expression of Oct3/4 was detected in the cytoplasm of CDCs from older mdx and wild-type mice (using antibodies from two suppliers), as has been seen in adipose stromal cells [33]. To investigate the effect of the increased cardiac fibrosis at 18 months of age, coexpression of CD90 and the cardiac fibroblast marker DDR2 was measured in CDCs from a separate set of wild-type and mdx mice (Fig. 3I). Although heart tissue from older mdx mice showed significantly more fibrosis than that from wild-type mice (Fig. 2C), there was no difference in expression of DDR2 between CDCs from wild-type or mdx mice. CD90 can be expressed on cardiac fibroblasts, however few cells that expressed both CD90 and DDR2 were detected. CDCs from 18-month-old mice were treated with 5-azacytidine and ascorbic acid for 2 weeks to induce differentiation. There were no significant differences in expression of cTnT or cMHC, measured using flow cytometry, between treated CDCs from wild-type and mdx mice (Fig. 4A, B), although the variability in differentiation was higher in the cells from mdx mice. Dystrophin expression was confirmed in differentiated CDCs from wild-type mice (Fig. 4B), however, although CDCs increased expression of cTnT and cMHC, the expression of these sarcomeric proteins, and of dystrophin, was diffuse and the cells did not resemble adult cardiomyocytes, suggesting that, in cells from older animals, treatment for 2 weeks resulted in only partial differentiation.
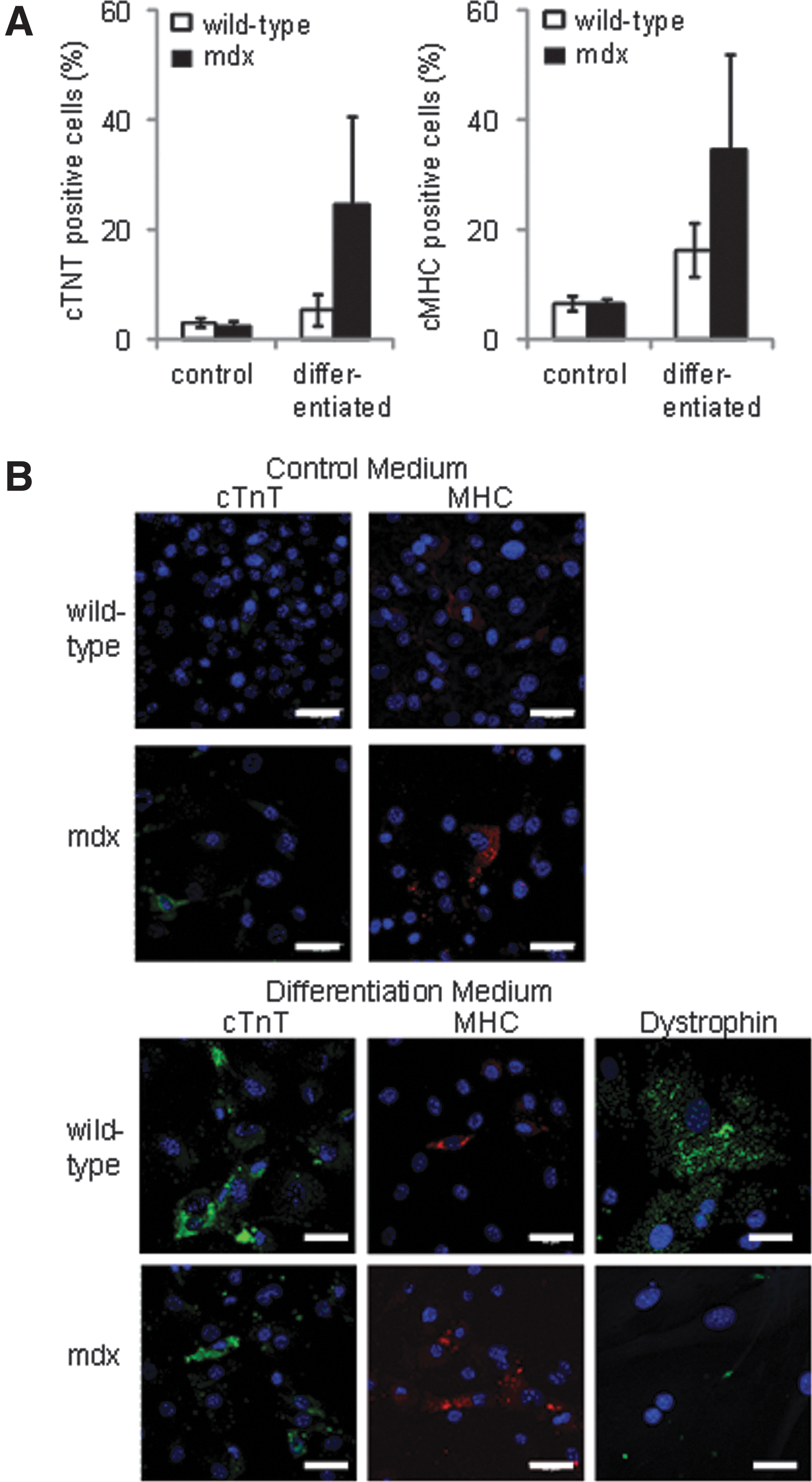
Differentiation of CDCs from older wild-type and mdx mouse hearts. There was no significant difference in expression of cardiac proteins, troponin T and myosin heavy chain in 5-azacytidine-treated CDCs from 18-month-old wild-type or mdx mice
Discussion
CDCs were isolated and cultured from hearts of C57Bl/10 mice aged 1.5, 6, 18, and 24 months. This is the first time that the effects of such a range of chronological ages on murine CDCs was investigated in age groups that are equivalent to teenage, adult, old, and very old humans. Explants were taken from the atria only, as these have the highest density of progenitor cells and it is thought that progenitors migrate from the atria to the ventricle in response to damage [34,35]. The results of the study clearly displayed an age-dependent reduction in the quantity of EDCs and CDCs and impairment of migration, clonogenicity, proliferation, and differentiation. We found that the numbers of EDCs and CDCs were significantly reduced from the older animals compared to the 1.5-month-old mice. It is possible that the increased fibrosis found in hearts from older animals may impair migration of the EDCs from within the explant tissue, thereby reducing cell numbers. Further, EDCs from aged animals had decreased migratory capacity and there was a substantial reduction in the proliferation rate of mouse CDCs in the aged groups, as has been reported for human CDCs [36].
Cells that migrate from explants include endothelial cells and epicardial progenitors and the myocardial progenitor cells. Mature myocardial cells do not survive long in culture and the proportion of adherent fibroblasts is reduced by the cardiosphere culture. Nevertheless, the CDC population is heterogeneous and includes cells expressing the pluripotent markers Oct3/4 and Sox 2 and spontaneously differentiating cells expressing smooth muscle actin and cTnT (Fig. 1E). The proportion of CSCs that expressed c-kit or Sca-1 was persistently downregulated in CDC populations with increasing age. This is consistent with a human CDC study, which confirmed that c-kit expression declined with advancing age ranging from neonates to teenagers [36]. Studies have shown a link between age and decreased ability to self-renew in various stem cell populations, such as endothelial progenitor cells [37] and neural stem cells [38]. In contrast, the proportion of cells expressing CD90 (Thy-1) increased with age, possibly due to a decline in other progenitors in aged CDC populations. CDCs from older mice contained a lower percentage of clonogenic cells compared with those from younger groups. Interestingly, Samper et al. found that hematopoietic stem cells from telomerase-deficient mice had a limited colony-forming capacity, suggesting that age-related downregulation of telomerase activity might result in impairment of clonogenicity [39]. Similarly, aging had a negative effect on CDCs' potential for cardiomyogenic differentiation. Differentiation of bone marrow-derived MSCs into adipocytes and chondrocytes is also adversely affected by age [40,41]. The degeneration in differentiation potential of stem cells could result from abnormal self-renewal due to telomere shortening and telomerase deficiency in aged stem cells indicated here and by others [42]. Telomerase, active in stem cells, delays telomere erosion and prevents chromosomal instability by elongating telomeres to protect against the loss of DNA during replication [43] and it is thought that the telomere-telomerase system contributes to age-related changes of stem cell phenotype [44]. Torella et al. found that the percentage of c-kit+ cells showing evidence of senescence, shortened telomere length and apoptosis was elevated in older wild-type mice [45].
The major function of dystrophin is to stabilize the sarcolemma during repeated cycles of contraction and relaxation of muscle cells [46,47]. In dystrophin-deficient hearts, the lack of dystrophin results in the loss of membrane integrity and increased susceptibility to damage from muscle contraction-induced stress, which in turn triggers a cascade of adverse downstream events, such as calcium overload and protein degradation, eventually leading to the death of cardiomyocytes [48]. Typically, the myocardial cell damage and death in DMD is limited to microinfarcts dispersed through the heart [49]. As the disease progresses, the ventricles gradually stretch and enlarge, which causes chamber dilation and wall thinning, leading to a reduction of contractility, dilated cardiomyopathy, and ultimately end-stage heart failure [47].
In a longitudinal study of mdx mice aged from 1 to 12 months using in vivo high-resolution MRI, we detected abnormalities in left ventricular function at 9 months and in RV function as early as 3 months [27]. Similarly, here, RV abnormality was found to precede left ventricular dysfunction, perhaps due to increased pressure load in the right ventricle, resulting from earlier onset of respiratory muscle weakness and pulmonary hypertension [27]. Further, ventricular dilation and dysfunction increased with age, in comparison to age-matched wild-type mice or young mdx mice. Dispersed focal areas of fibrosis were seen in right and left ventricles of mdx mouse hearts but not in wild-type mouse hearts. Necrotic lesions have been found in the hearts of mdx mice as early as 6 weeks [50], but the significant difference in the amount of fibrocollagenous tissues between control and mdx mice does not occur until 6 months of age [27] and Quinlan et al. reported that patchy fibrosis affects all regions of the left ventricle and the right ventricle to an approximately equal extent in 17-month-old mdx mice [22]. In contrast to their findings, our data show that the percentage of scar tissue in the right ventricle was significantly higher than that in the left ventricle (P<0.05), but the degree of fibrotic change was similar in distinct left ventricular areas in the mdx mice at both 6 and 18 months. However, we found that CDCs could be generated from the atrial tissue of mdx mice aged 6 and 18 months. Similar to wild-type control mice, EDC, cardiosphere, and CDC yield from old mdx mice was significantly less than that from young mdx mice, but we did not detect substantial differences in CDC number or characteristics between cells from mdx mice and wild-type mice of either age. However, nuclear expression of Oct3/4 and Sox2 was not seen in CDCs from older mdx mice, suggesting that these cells were somewhat less stem-like than those from age-matched wild-type mice, and that the combined effects of age and damage in the dystrophic heart may have depleted the most stem-like progenitors in the atrial niche. It may be that, although there is an inflammatory response to the cell necrosis caused by contraction-induced stress, this does not induce the level of ROS seen in ischemic heart damage. Second, the damage to the myocardium occurs gradually, at small focal locations through the heart, so that stem cell pool may be maintained through self-renewal. The CDCs were isolated from the atria, which have been reported to be the most abundant region of functionally competent CSCs [36,51], and, in DMD, fibrotic scarring generally spares the atria [52], likely due to the reduced mechanical stress in this region. However, we had hypothesized that the progenitor population in the atria would be depleted by an increased requirement for cells to repair microinfacts in the ventricle. Cassano et al. demonstrated that ventricle-derived CPCs from golden retriever muscular dystrophy (GRMD) dogs showed impaired self-renewal and cardiomyogenic differentiation in vitro and in vivo when compared to those from healthy dogs [53]. In comparison, a recent study showed that there were no obvious alterations to cell biology observed between satellite cells isolated from wild-type or GRMD dystrophic dogs, as determined in vitro by proliferation capacity, clonogenicity, motility, and expression of terminal differentiation proteins [54].
In summary, we have demonstrated that CDCs can be isolated from mouse hearts as old as 24 months but that CDCs numbers and regenerative potential decline with age. Although it is known that CPC numbers are adversely affected by an acute ischemic insult [55], here we demonstrate that, when cardiac damage occurs slowly and progressively, the CPC population is maintained.
Footnotes
Acknowledgment
This work was supported by the British Heart Foundation (grant nos. PG/07/059/23259 and RG/07/004/22659).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.