
Abstract
There are well-established approaches for osteogenic differentiation of embryonic stem cells (ESCs), but few show direct comparison with primary osteoblasts or demonstrate differences in response to external factors. Here, we show comparative analysis of in vitro osteogenic differentiation of mouse ESC (osteo-mESC) and mouse primary osteoblasts. Both cell types formed mineralized bone nodules and produced osteogenic extracellular matrix, based on immunostaining for osteopontin and osteocalcin. However, there were marked differences in the morphology of osteo-mESCs and levels of mRNA expression for osteogenic genes. In response to the addition of proinflammatory cytokines interleukin-1β, tumor necrosis factor-α, and interferon-γ to the culture medium, primary osteoblasts showed increased production of nitric oxide (NO) and prostaglandin E2 (PGE2) at early time points and decreases in cell viability. In contrast, osteo-mESCs maintained viability and did not produce NO and PGE2 until day 21. The formation of bone nodules by primary osteoblasts was reduced markedly after cytokine stimulation but was unaffected in osteo-mESCs. Cell sorting of osteo-mESCs by cadherin-11 (cad-11) showed clear osteogenesis of cad-11+ cells compared to unsorted osteo-mESCs and cad-11− cells. Moreover, the cad-11+ cells showed a significant response to cytokines, similar to primary osteoblasts. Overall, these results show that while osteo-mESC cultures, without specific cell sorting, show characteristics of osteoblasts, there are also marked differences, notably in their responses to cytokine stimuli. These findings are relevant to understanding the differentiation of stem cells and especially developing in vitro models of disease, testing new drugs, and developing cell therapies.
Introduction
D
In vivo, bone development is highly regulated and results in an organized and hierarchically ordered structure [6]. Bone development progresses through distinct developmental stages starting with the commitment of mesenchymal stem cells (MSCs) to the osteoblast lineage, proliferation of osteoprogenitors, and maturity of the differentiated osteoblast, leading to the formation of mineralized extracellular matrix (ECM) [7]. To produce osteoblasts effectively from ESCs, this progression needs to be followed in vitro.
In vitro differentiation of osteoblasts results in the formation of distinct colonies of mineralized bone-like matrix, known as bone nodules [8,9]. The ECM deposited by osteoblasts in vitro has been shown to include collagen-I (col-I), fibronectin, osteocalcin (OCN), and osteopontin (OPN), and staining for these proteins is often most predominant around the mineralized nodules [10 –13]. The process of osteogenesis is coordinated by various transcription factors, with Runx2 and osterix being regarded as key regulators [14 –16].
Both mouse [17,18] and human ESCs [19 –21] have been shown to display the features of osteogenically differentiated cells in vitro, exhibiting molecular and structural features resembling bone tissue by the formation of mineralized bone nodule structures. The majority of osteogenic protocols for ESCs direct cell differentiation by including factors in the culture medium, such as β-glycerophosphate (BGP), ascorbate, dexamethasone, simvastatin, retinoic acid, vitamin D3, and bone morphogenic proteins [3,22 –30]. Although traditional osteogenic differentiation strategies for ESCs leads to the formation of bone nodules and expression of osteogenic markers, little research has compared this to the in vitro differentiation of osteoblasts.
Osteogenic differentiation is often shown by the presence of osteogenic markers, but it is also useful to explore the functional biochemical response of the cells to certain stimuli, in comparison to osteoblasts. In this study, we examine the responses of the cells to cytokines associated with inflammation, including interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and interferon-γ (IFN-γ). These proinflammatory cytokines are proteins that co-ordinate local and systemic inflammation and have in vitro effects on osteoblast proliferation, collagen synthesis, mineralization, and alkaline phosphatase (ALP) activity [31 –35]. Responses to proinflammatory environments can be measured by increased prostaglandin E2 (PGE2) and nitric oxide (NO), changes in cell viability, and expression of inducible enzymes [36,37]. The response of osteoblasts to proinflammatory cytokines has been investigated extensively [31 –38], whereas little work has been performed on ESC-derived osteogenic cells. The impact of inflammation in osteogenic differentiation may also be of some importance when producing a potential cell therapy. A regenerative medicine product would be manufactured under favorable conditions, supporting cell growth and viability. Subsequently subjecting it to a damaged/diseased environment could have a significant effect on the success or failure of the final therapy.
To progress ESCs to use in cell therapies and regenerative medicine, differentiation needs to be fully validated, and it is likely that a cell selection step will be required to isolate a purified cell population of interest. Currently, there is no commonly used cell surface marker of the early osteoblast for cell sorting. In this study, we investigate cadherin-11 (cad-11) as a marker for the purification of osteogenically differentiated ESCs. Cad-11 has previously been used to purify ESCs [18] and is a cell adhesion molecule strongly associated with bone formation and osteogenic differentiation [39,40].
In this study, we compare the osteogenic differentiation of mouse ESC (mESC) with mouse primary osteoblasts and subsequently study the differentiation of a subpopulation of ESC selected for cad-11 expression. We describe significant differences in the osteogenic differentiation of the cell types and show significant differences in responses of the cell types to exposure to the proinflammatory cytokines IL-1β, TNF-α, and IFN-γ.
Materials and Methods
Cell culture
Mouse ESCs (CEE line [41]) were cultured on a monolayer of mitotically inactivated mouse fibroblasts (SNL line), as previously described [42]. Cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% fetal bovine serum (FBS; Biosera), 100 U/mL penicillin and 100 μg/mL streptomycin (Sigma), 2 mM
Primary calvarial osteoblasts were obtained from 1- to 3-day-old CD1 mouse calvaria by sequential enzymatic digestion. Calvaria were dissected and digested in a solution of 1.4 mg/mL collagenase type IA (Sigma) and 0.5 mg/mL trypsin II S (Sigma). Cells released during the first two populations (10 min digestions) were discarded, and populations of cells from the next three digestions (20 min digestions) were plated in tissue culture flasks at a density of 6.6×103 cells/cm2. Cells were cultured in minimum essential medium-α (Lonza), containing 10% FBS (Sigma), 100 U/mL penicillin and 100 μg/mL streptomycin, and 2 mM
Osteogenic differentiation of mESCs
Embryoid bodies (EBs) were formed from mESCs by mass suspension, in mESC culture medium without LIF for 3 days. Trypsin-mediated dissociation of EBs, yielded a single cell suspension that was plated at a density of 10,600 cells/cm2 in 0.1% (w/v) gelatin-coated well plates. Osteogenic differentiation was induced by supplementing the medium with 50 mM BGP, 50 μg/mL ascorbate 2-phosphate, and at points when proinflammatory cytokines were not present, 10 μM dexamethasone. From this point forward, mESCs were referred to as osteogenically differentiates mESCs (OSTEO-mESCs) and treated identically to primary osteoblasts.
Alizarin red S staining
Cell monolayers were fixed in 10% (v/v) formalin and stained for the presence of calcified matrix using a 2% (w/v) solution of alizarin red S (pH 4.2; Sigma) for 4 min at room temperature. Images were taken with a Nikon Eclipse 90i stereo dissection microscope or Nikon Eclipse TS100 inverted microscope with Hoffman contrast. Image analysis and quantification of area stained was performed using ImageJ version 1.43U (NIH).
Immunocytochemistry
Cell culture plates were fixed with a 4% (w/v) solution of paraformaldehyde (Sigma) for 20 min. After washing with phosphate-buffered saline (PBS), cells were permeabilized with 0.1% (v/v) Triton X-100 (Sigma) for 45 min and washed again with PBS. After blocking nonspecific binding by incubating in 3% (v/v) donkey serum (Sigma) for 20 min, cells were incubated with primary antibodies at 4°C overnight. After washing with PBS, cells were incubated with secondary antibodies (donkey Alexa-Fluor-488 or 546; Invitrogen) for 1 h at room temperature. Nuclear counterstaining with Hoechst 33258 was performed. Details of antibodies can be found in Supplementary Table S1 (Supplementary Data are available online at
Real-time quantitative polymerase chain reaction
RNA was extracted using an RNeasy kit (Qiagen) according to the manufacturer's instructions. Reverse transcription of 400 ng RNA into single-stranded cDNA was performed using the superscript II system (Invitrogen), according to the manufacturer's instructions. Real-time quantitative polymerase chain reaction (RT-qPCR) was performed on a MyiQ RT-PCR detection system (Bio-Rad) with iQ SYBR Green supermix (Bio-Rad). Efficiency and threshold cycle (CT) calculations were performed using online software based on a four-parameter simple exponent model [43]. Expression levels were calculated using an efficiency-corrected comparative threshold cycle method (EΔΔCT). All values were normalized to the Rpl32 ribosomal protein gene. Primer sequences were as follows: Rpl32 (Fwd) TTAAGCGAAACTGGCGGAAAC, (Rvs) TTGTTGCTCCCATAACCGATG; Runx2 (Fwd) TGTTCTCTGATCGCCTCAGTG, (Rvs) CCTGGGATCTGTAATCTGACTCT; Col1a1 (Fwd) GCTCCTCTTAGGGGCCACT, (Rvs) CCACGTCTCACCATTGGGG; Opn (Fwd) AGCAAGAAACTCTTCCAAGCAA, (Rvs) GTGAGATTCGTCAGATTCATCCG; Ocn (Fwd) CTGACCTCACAGATGCCAAGC, (Rvs) TGGTCTGATAGCTCGTCACAAG.
Proinflammatory cytokine treatment
Recombinant human IL-1β, human TNF-α, and mouse IFN-γ (R&D Systems) were added to the culture medium at a concentration of 1, 10, and 100 ng/mL, respectively, unless otherwise stated. IL-1β and TNF-α have cross species reactivity between mouse and human, and IFN-γ does not possess this property. Due to the anti-inflammatory properties of dexamethasone, this was removed from the osteogenic medium when the proinflammatory cytokines were present.
Nitrite and PGE2 production
Nitrite and PGE2 in the culture medium were measured after 48 h of proinflammatory cytokine treatment at various stages of osteogenic culture. Nitrite was measured as an estimation of NO production, using the Griess Reagent System (Promega) according to the manufacturer's protocol. Briefly, 50 μL of sample was transferred to a 96-well plate, 50 μL of 1% sulphanilamide added, and incubated for 5 min at room temperature, before 50 μL 0.1% N-(1-naphthyl)ethylenediamine (NED) added and incubated at room temperature. Absorbance was measured at 540 nm, with correction at 690 nm, using a plate reader (TECAN Infinite 200 PRO). Concentration calculated from standard curve.
PGE2 concentration within the culture medium was determined by enzyme immunoassay (EIA) using a commercially available kit (Parameter™ PGE2 Assay; R&D Systems), according to the manufacturer's protocol. Briefly, a competitive binding EIA was performed on the threefold diluted samples. Optical absorbance was read at 450 nm with correction at 570 nm. PGE2 in the sample was determined using standard curves. Both nitrite and PGE2 values were corrected for DNA content of formalin-fixed cells, determined by fluorescent Hoechst readings, at excitation wavelength of 360 nm and emission wavelength of 460 nm.
Cell viability assay
Cells were plated in 96-well plates. A dose response of IL-1β, TNF-α, and IFN-γ was performed by keeping the cytokines at a ratio of 1:10:100, but performing a serial dilution to lower the total dose. Measurements of viability were taken using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS assay; Promega) at certain time points, as per the manufacturer's protocol. Briefly, 20 μL MTS reagent was added to each cell-containing well, in 100 μL culture medium. Cells were incubated at 37°C for 1 h before absorbance read at 490 nm with wavelength correction at 690 nm.
Alkaline phosphatase assay
After osteogenic culture, cell monolayers were lysed in a solution of 0.1% (v/v) Triton X-100 in 0.2 M Tris buffer and cell layer was collected using a cell scraper. Samples were centrifuged at 10,000 g for 10 min and transferred to a 96-well plate. ALP activity was assessed by the addition of 1 mg/mL p-nitrophenyl phosphate (pNPP) solution in 0.2 M Tris buffer (Sigma). After 20 min incubation at room temperature, optical absorbance was read at 405 nm. ALP concentration was determined from a standard curve.
Magnetic-activated cell sorting
Osteo-mESCs were cultured on gelatin-coated flasks in the osteogenic medium for 16 days. Cells were removed from flasks and dissociated by trypsin/EDTA treatment, and the cell suspension passed through a 70-μm cell strainer to remove matrix components. Cells were resuspended in cad-11 primary antibody (R&D Systems) at 10 μg/mL for 5 min at room temperature, incubated in biotinylated secondary antibody (Vector) for 10 min at 4°C, and finally, incubated with antibiotin magnetic microbeads (Miltenyi Biotec). Cells were sorted for cad-11 expression using the MiniMACS™ separator in combination with MS columns (Miltenyi). Both cad-11+ and cad-11− osteo-mESCs were plated at a density of 20,000 cells/cm2 in gelatin-coated well plates and cultured in the osteogenic medium.
Statistical analysis
Statistical significance between the groups was analyzed using PASW Statistics 18.0.3 software. Two groups were compared by unpaired Student's t-test, and multiple groups were compared by one-way ANOVA.
Results
Osteogenic differentiation of mESCs compared to primary osteoblasts
In vitro osteogenic differentiation of mESCs was compared to the differentiation of mouse primary osteoblasts (Fig. 1). After 21 days in the osteogenic medium, both cell types showed the formation of specific areas of calcified matrix or bone nodules, revealed by alizarin red staining (Fig. 1A). Subjective observation showed nodules were more abundant in osteo-mESC cultures. Non-osteogenic controls showed little evidence of staining, although some diffuse nonspecific background staining of the osteo-mESC matrix was present.
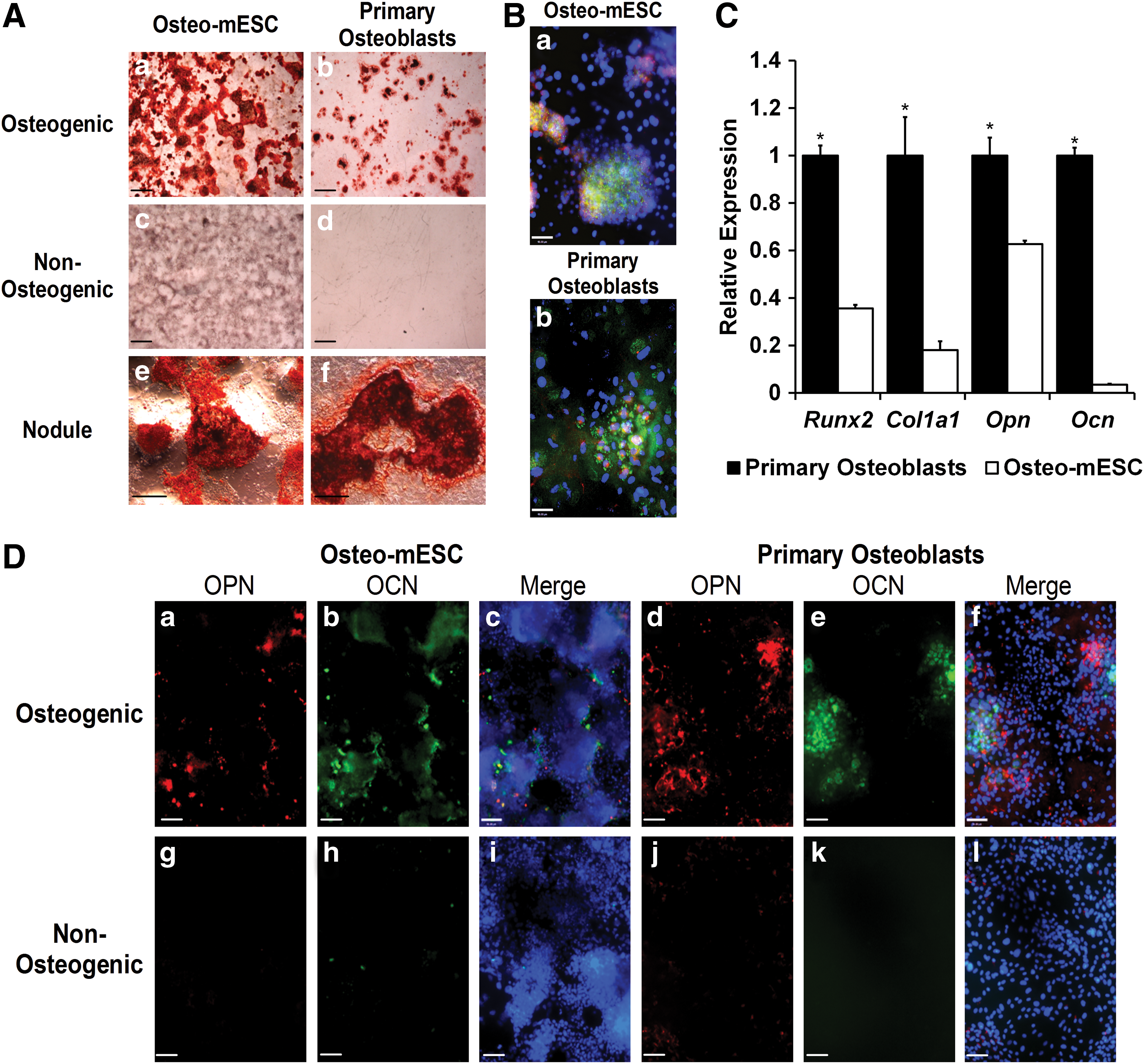
Osteogenic differentiation of mouse embryonic stem cells (osteo-mESCs) and mouse primary calvarial osteoblasts.
Immunocytochemistry at day 28, revealed staining for col-I and cad-11 (Fig 1B) and OPN and OCN (Fig. 1D) within and around bone nodules formed by both cell types. Staining of OPN and OCN in the non-osteogenic medium was minimal. Visually, there were noticeably more cells present in osteo-mESC cultures, and cells were smaller and more densely packed, as shown by Hoechst stain.
RT-qPCR analysis of expression of Runx2, Col1a1, Opn, and Ocn at day 18 of osteogenic culture (Fig. 1C) demonstrated significantly lower relative expression levels of all four genes in osteo-mESCs, particularly OCN, which is a late marker of osteogenesis.
Response of osteo-mESCs to proinflammatory cytokine treatment compared to primary osteoblasts
Preliminary work was performed investigating the response of the osteo-mESCs and primary osteoblasts to individual cytokines and different combinations of cytokines. Nitrite (Supplementary Fig. S1) and PGE2 (Supplementary Fig. S2) release shows that significant responses were only seen from the primary calvarial cells when two or more cytokines were combined and were only seen from the osteo-mESCs when three cytokines were combined during the later stages of the osteogenic culture. From these results, it was decided to use all three cytokines in combination to achieve the maximum response.
Osteo-mESCs at different time points of culture and osteoblasts at day 7 were exposed to IL-1β, TNF-α, and IFN-γ by addition to the culture medium for 48 h. Nitrite (Fig. 2A), PGE2 (Fig. 2C), and expression of inducible nitric oxide synthase (iNOS), the enzyme that leads to NO production, and cyclooxygenase-2 (COX-2) and prostaglandin E synthase (PGES), the enzymes leading to PGE2 (Fig. 2B), were assessed. In response to cytokines, osteoblasts produced large amounts of nitrite and PGE2. In comparison, osteo-mESCs did not produce PGE2 in response to cytokine stimulation until day 14 of osteogenic differentiation. Osteo-mESCs did not produce significant concentrations of nitrite, until day 21. In comparison to the 7-day cultured osteoblasts, levels produced were lower at all the time points. To corroborate nitrite and PGE2 results, staining for iNOS, COX-2, and PGES was performed (Fig. 2B). Osteoblasts showed positive staining for iNOS, COX-2, and PGES in the majority of cells (Fig. 2B-a, f, l). Osteo-mESCs did not show any staining for iNOS in response to cytokines until day 21, and this was clustered around areas of dense nuclear staining. Both COX-2 and PGES staining was seen when cytokine-treated at day 14 and 21, in accordance with the PGE2 results. No staining was seen in unstimulated controls (data not shown).
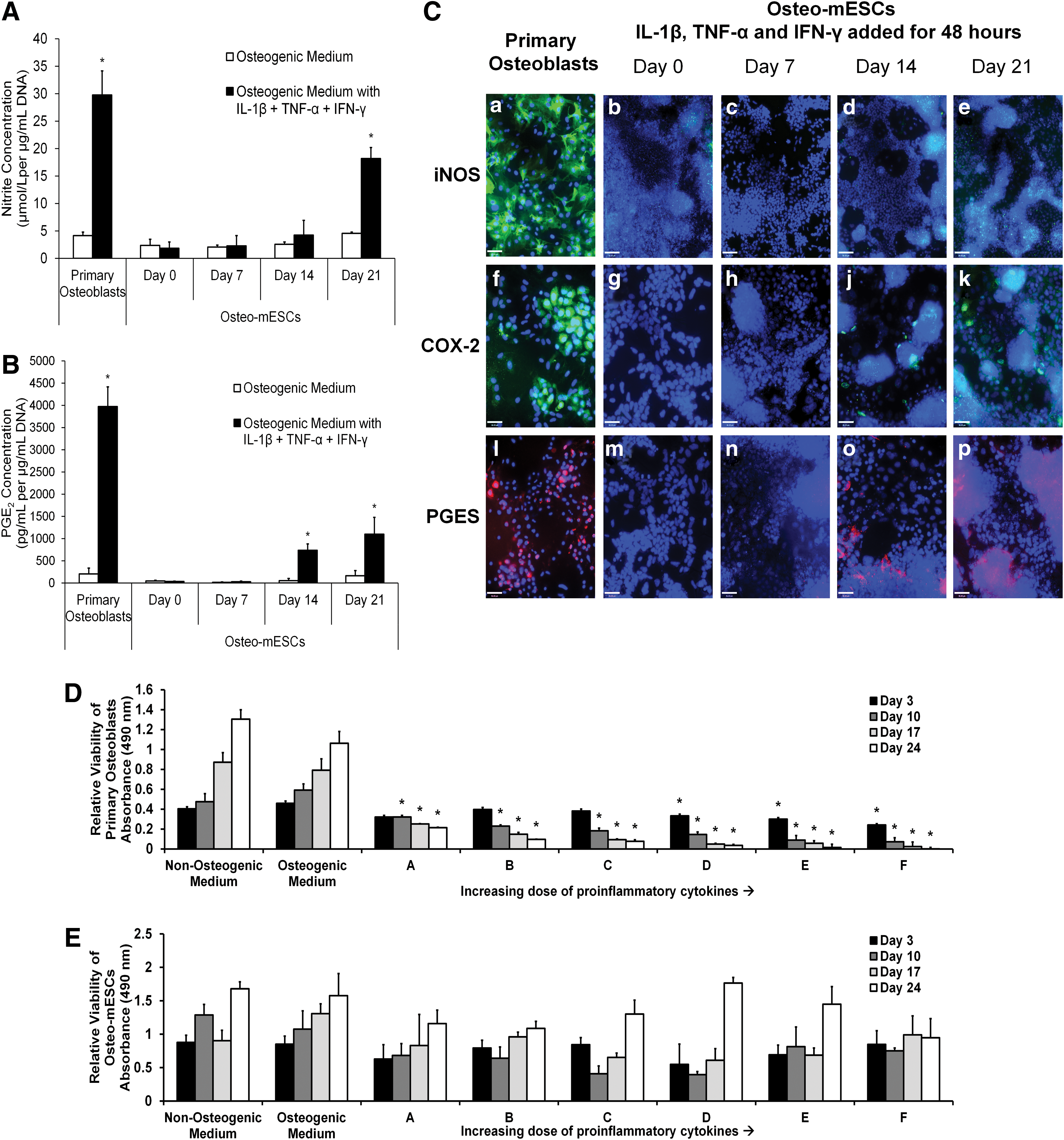
Response of osteo-mESCs to proinflammatory cytokine stimulation compared to primary osteoblasts.
Changes in cell viability in response to IL-1β, TNF-α, and IFN-γ treatment was assessed by MTS assay; cells were treated with cytokines continually over a 24-day period at a range of cytokine concentrations, all with ratio 1:10:100 (IL-1β:TNF-α:IFN-γ; Fig. 2D, E). Primary osteoblasts showed significant reductions in viability at all cytokine concentrations from the lowest (A: 0.03125 ng/mL IL-1β, 0.3125 ng/mL TNF-α, and 3.125 ng/mL IFN-γ) to the highest concentration (F: 1 ng/mL IL-1β, 10 ng/mL TNF-α, and 100 ng/mL IFN-γ). In comparison, osteo-mESCs showed no deleterious changes in cell viability over time, at any cytokine concentration (Fig. 2E).
Effect of proinflammatory cytokines on bone nodule formation
To assess the effects of short bursts of cytokine stimulation on the osteogenic differentiation of osteo-mESCs and primary osteoblasts and form bone nodules, cells were stimulated with IL-1β, TNF-α, and IFN-γ at day 0, 3, 7, or 14 of osteogenic culture for 48 h, before being returned to control osteogenic medium. Alizarin red staining (Fig. 3A) and subsequent quantification of staining (Fig. 3E, F) demonstrated that the cytokines had little effect on bone nodule formation of osteo-mESCs when applied on day 0 or 3. When applied on day 7, the proportion of staining relative to the control was significantly higher. In contrast, when stimulated on day 14, levels of alizarin red staining were significantly lower. Effects of cytokine stimulation were more apparent in the formation of nodules by primary osteoblasts. No staining was seen when cells were stimulated on day 0, 3, or 7, and only small nodules formed after stimulation on day 14, in comparison to unstimulated control.
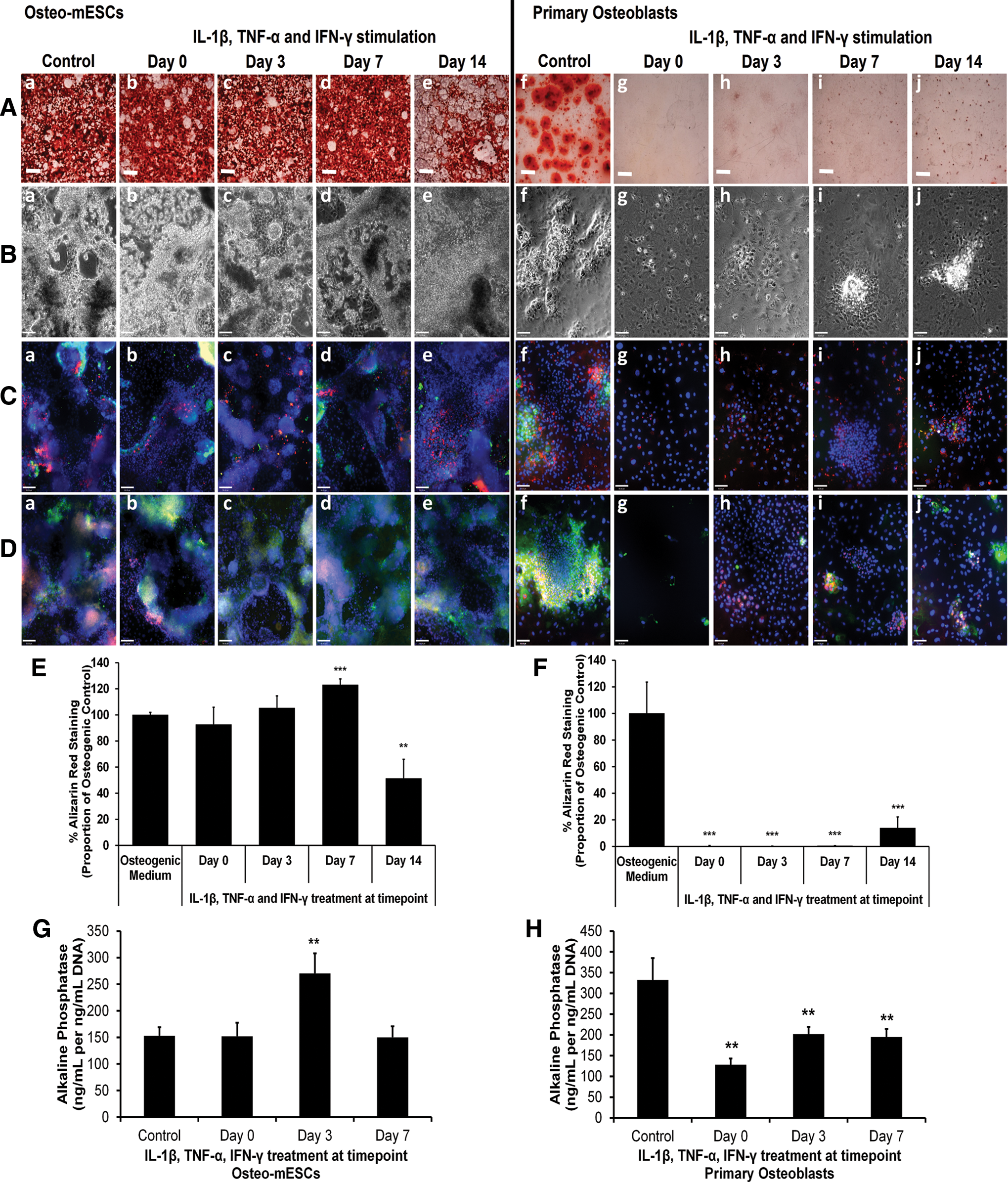
Effect of IL-1β, TNF-α, and IFN-γ on bone nodule formation of osteo-mESCs and primary osteoblasts. Cells were stimulated at day 0, 3, 7, or 14 with IL-1β, TNF-α, and IFN-γ for 48 h, before being returned to the osteogenic medium and cultured continuously until either day 14 (alkaline phosphatase) or day 21.
Phase-contrast images of the osteo-mESCs (Fig. 3B-a–e) showed heterogeneous areas of cells with considerable matrix production. Images of primary osteoblasts revealed the formation of obvious raised nodules in control cultures (Fig. 3B-f), which were not present in cells stimulated on day 0 or 3 and considerably smaller when stimulated on day 7 and 14. Immunocytochemistry of OCN, OPN, col-I, and cad-11 (Fig. 3C, D) demonstrated that short bursts of cytokine stimulation disrupted ECM deposition of the primary osteoblasts at all the time points. Conversely, osteo-mESCs continued to produce ECM proteins and express cad-11, in nodular areas, despite cytokine stimulation.
Effect of cad-11 sorting on osteo-mESC differentiation
Osteo-mESCs were cultured until day 16 in osteogenic medium and subsequently sorted based on expression of cad-11 using magnetic-activated cell sorting (MACS). Average percentage of cad-11-positive cells was 21.2%, and numbers were comparable between cell sorting experiments (standard deviation 3.88, n=6). Cad-11+ and cad-11− cells were cultured in the osteogenic medium for up to a further 21 days. Proinflammatory cytokine stimulation was performed in a short burst of 48 h at day 7 and 14 post-sorting, as described previously with nonsorted cells. Alizarin red staining at 21 days (Fig. 4A) indicated cad-11+ cells formed discrete nodules, similar to the nodules formed by primary osteoblasts but appeared unaffected by cytokine stimulation. Cad-11− osteo-mESCs did not form nodules and only showed light alizarin red staining but was also unaffected by cytokine stimulation. Phase-contrast images (Fig. 4B) further demonstrated that nodules were formed by cad-11+ cells and revealed the different morphology of the cad-11− cells. Cad-11+ osteo-mESCs stained positive in nodular areas for OCN and OPN (Fig. 4C-a–c) and col-I (Fig. 4D-a–c). Conversely, cad-11− cells did not stain for OPN and lightly stained for OCN (Fig. 4C-d–f). Col-I staining was very sparse (Fig. 4D-d–f). Staining of neither cad-11+ or cad-11− osteo-mESCs was affected by the proinflammatory cytokine stimulation, at either time point.
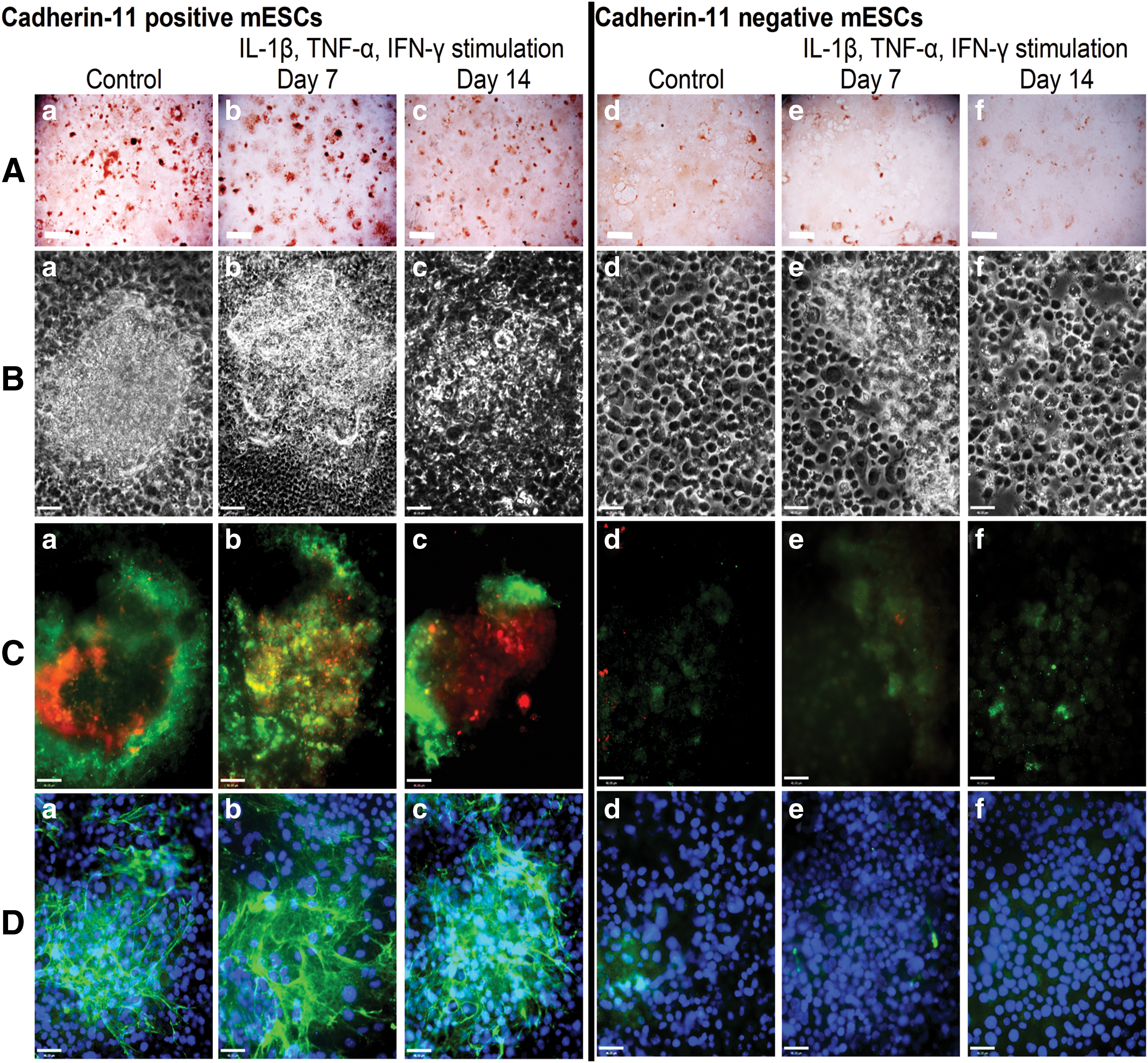
Osteogenic differentiation of cad-11 sorted osteo-mESCs and effect of proinflammatory cytokines on bone nodule formation. Cells were sorted for cad-11 at day 16 of osteogenic culture and positive and negative fractions cultured separately in osteogenic medium. Cells were stimulated with IL-1β, TNF-α, and IFN-γ for 48 h, at day 7 or 14 post-sorting before being returned to the osteogenic medium and cultured until day 21.
Response of cad-11 sorted osteo-mESCs to proinflammatory cytokines
To assess further differences between cad-11+ and cad-11− osteo-mESCs, response of cells to IL-1β, TNF-α, and IFN-γ, in terms of nitrite production (Fig. 5A, B), PGE2 production (Fig. 5C, D), and expression of iNOS (Fig. 5E) was assessed at day 7 and 14 post-sorting. Both cad-11+ and cad-11− cells produced significant amounts of nitrite and PGE2 in response to the cytokines at both day 7 and 14. However, cad-11+ cells produced significantly more nitrite than cad-11−. This was also reflected in iNOS staining; almost all cad-11+ cells stained for iNOS, but few cad-11− cells stained positive, suggesting a heterogeneous population. Unstimulated control cultures did not stain for iNOS, in all cases. Stimulated cad-11− cells produced significantly more PGE2 at day 7 than cad-11+, but this was also true of the cad-11− control medium. Production of PGE2 was subsequently reduced by day 14, where it was significantly lower than the cad-11+ cells. Levels of nitrite and PGE2 produced in response to the cytokines were of a similar magnitude to those produced by primary osteoblasts at day 7 (Fig. 2).
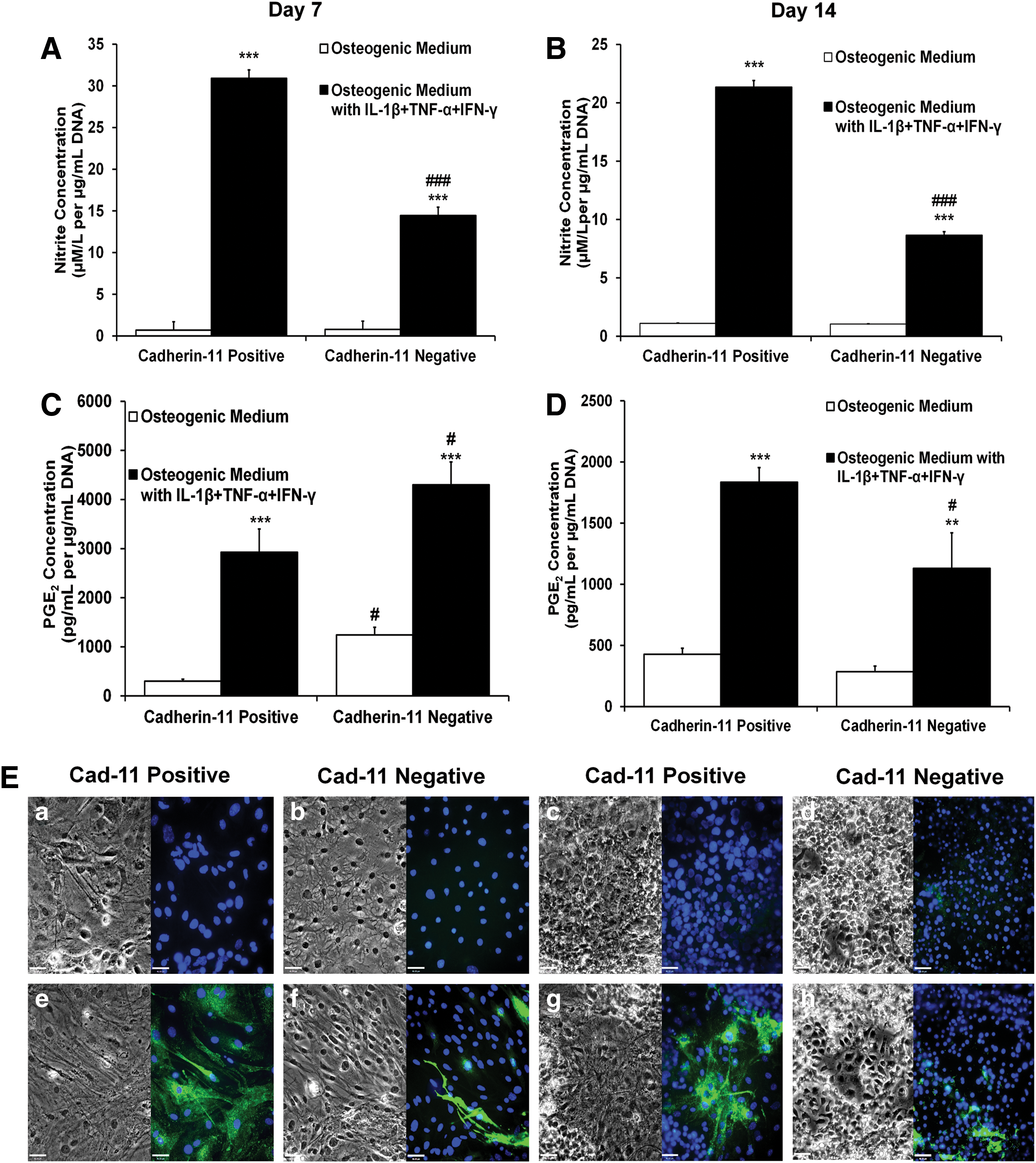
Response of cad-11 sorted cells to proinflammatory cytokine stimulation.
Discussion
In vitro osteogenic differentiation of primary osteoblasts [8 –13,44] and ESCs [17 –21] have both been well described, but there have been few comparative studies. In this study, both cell types showed expression of markers indicative of osteogenic differentiation and formed nodules comprising mineralizing matrix. Both cell types stained for proteins associated with osteoblast differentiation such as OPN, OCN, and col-I, particularly associated with the cell colonies/bone nodules. Expression of the cell-to-cell adhesion molecule cad-11, found in high levels on osteoblasts, and important in the formation of mesenchymal tissues in embryo development [45,46], was also seen in both cell types. Despite these similarities, the morphology of the osteo-mESC cells was noticeably different, showing a heterogeneous population and larger numbers of nuclei within nodules. Formation of bone nodules within cultures subjectively appeared different when imaged at both low and high magnification. Despite, the formation of more bone nodules in the osteo-mESCs, there was a lower expression of osteogenic genes. This can be explained by nonspecific binding of calcium within the osteo-mESC matrix, which may not be related to osteogenesis.
There is certainly interest in the comparison of osteoblasts derived from primary and stem cell sources [47 –49]. Gentleman et al. [47] compared mineralized nodules of neonatal mouse osteoblasts, bone marrow-derived-MSCs, and mESCs. They concluded that the bone nodules formed by mESCs showed distinct differences to those of the osteoblasts and MSCs, in terms of formation time and Raman spectra of mineral structure [47]. The results of our study support this, as we saw morphological differences in the formation of bone nodules and expression of ECM proteins. We also provide additional evidence that mESCs do not have the same responses to proinflammatory cytokines as primary osteoblasts, indicating a difference in biochemical response. Although both cell types form bone nodules by day 21, the response of the osteo-mESCs to cytokine stimulation was very different to that of primary osteoblasts, which showed reduced cell viability and marked production of nitrite and PGE2, and is consistent with the literature [37,38,50 –55]. In contrast, the osteo-mESCs showed no reductions in cell viability in response to extended cytokine stimulation nor any significant increases in the production of nitrite or PGE2, until the latter stages of the culture period at day 21; the time point at which traditional differentiation protocols indicate as fully differentiated. As well as revealing differences between cells types, investigation of the effects of proinflammatory cytokines on cell response, particularly ESCs, can give an indication of how cell therapies may perform when subjected to the environment of disease and inflammation into which they will be implanted. This response may have a significant bearing on the success or failure of the final therapy.
The effect that short bursts of IL-1β, TNF-α, and IFN-γ treatment had on the eventual osteogenic differentiation of primary osteoblasts and osteo-mESCs further suggested that the differentiation process of the ESC is not equivalent to that of primary osteoblasts. The osteoblasts showed marked responses to the transient exposure of proinflammatory cytokines, which had knock-on effects on the final differentiation state, with the formation of fewer and smaller bone nodules. When treated on day 0 or 3, cell viability fell during the 48 h in the presence of the cytokines and continued to fall after returning to the osteogenic medium with no cytokines, reducing final cell numbers significantly. IL-1β, TNF-α, and IFN-γ, have previously been shown to cause apoptosis of osteoblasts, particularly when applied in combination [51,53,56], this may have caused many of the eventual effects seen on cell differentiation.
Previous reports have focused on the effects of individual cytokines on osteoblast differentiation; but when used in combination, the effects are synergistic and enhanced [57,58]. IL-1β, TNF-α, and IFN-γ have been shown to inhibit bone nodule formation, decrease ALP activity, inhibit OCN and col-I deposition, in both mouse and human osteoblasts [33,35,59 –66]. Many of these effects are mediated by NO and PGE2 pathways, which are stimulated by these cytokines [32,67]. That these effects are not seen with the osteo-mESCs indicates, once again, that the in vitro differentiation process is different from that of primary osteoblasts.
The mechanisms that contribute to the differences in response of the osteo-mESCs to proinflammatory stimuli remain to be determined, but we can hypothesize that the receptor and signal transduction pathways may be less developed or less active in mESC-derived osteoblasts. ESCs and early ES-derived vascular cells have been shown to have a low level of TNF-receptors, and it has been reported that nuclear factor-κB, a transcriptional regulator that plays a key role in immunity and inflammation, has relatively low expression in undifferentiated ES cells but increases during differentiation [68,69]. Expression of Toll-like receptors, a group of receptors associated with the immune and inflammatory response, have also been shown to be significantly downregulated in hESCs [70].
To investigate further the distinct responses of the different cell types, we performed cell-sorting studies on the ESC cultures. There is no definitive well-characterized marker of the early osteoblast. Osteogenesis of ESCs often requires first inducing ES-derived MSCs [71,72], but the intention of this study was to avoid the need to perform this step and directly harvest a population of osteogenic cells from the mESC cultures. Cad-11 has previously been used to sort osteogenic cells within differentiating ESC-derived cultures [18]. In our cell sorting experiments, we achieved a cad-11-positive population of about 21%, which was considerably less than previously described [18]. It is worth noting that during the MACS process, there was a substantial reduction in the number of cells, and after seeding, many of the cells did not attach to the plate.
Cad-11+ cells differentiated to osteoblasts as suggested by the formation of bone nodules and immunostaining for OCN, OPN, and col-I, similar to primary calvarial cells. The cad-11− population was more heterogeneous, with little evidence of bone nodule formation or immunostaining. The advantage of using cad-11, as opposed to a marker such as STRO-1, is the ability to identify osteoprogenitors, rather than mesenchymal progenitors, and the level of characterization the cell surface marker has previously received [39,46,73,74].
Differentiation of the cad-11+ mESCs was unaffected by transient exposure to proinflammatory cytokines, in contrast to primary osteoblasts, which responded with reduced cell viability and nodule formation. This indicates that although comparable to osteogenic differentiation of primary osteoblasts, the cad-11+ cells were still distinct in biochemical response. However, unlike the unsorted osteo-mESCs, both cad-11+ and cad-11− cells responded to the presence of the cytokines by producing NO and PGE2, in similar amounts to the primary calvarial osteoblasts. It is also interesting to note that at 14 days post-sorting, levels of both NO and PGE2 production were lower than at 7 days. This decrease in production with progressive differentiation also occurred with the primary osteoblasts (data not shown). In cad-11+, iNOS staining was seen in most cells, whereas in cad-11− cultures, iNOS was seen only in a few cells, suggesting that a heterogeneous environment. Many cell types, such as fibroblasts and epithelial cells [75 –77], respond to the presence of these cytokines with the production of NO and PGE2, and it may be that the cad-11− mESCs were differentiated into cells other than the osteoblast. These results taken together suggest that in sorting for cad-11, a population of ESC-derived cells that responds more similarly to the early osteoblast was purified.
In this study, we demonstrate that traditional protocols for osteogenic differentiation of ESCs result in significant differences compared to the osteogenic differentiation of primary osteoblasts. In addition, we show significant differences in the response of osteogenic mESCs to a proinflammatory environment when compared to primary osteoblasts, indicating a dissimilar differentiation state. The findings of this study highlight the need for further investigation into osteogenic differentiation protocols, particularly a cell-sorting step. We demonstrated by selecting for cad-11+ cells, comparable osteogenic differentiation, with respect to primary osteoblasts. In order for osteogenically differentiated ESCs to progress to a clinical setting, differentiation protocols must be validated and establishment of a pure population of early osteoblasts must be demonstrated.
Footnotes
Acknowledgments
The authors thank Professor Jane Aubin and Frieda Chen at the University of Toronto for advice, assistance, and the kind provision of primer sequences. We also thank staff at the Biomedical Services Unit, University of Nottingham for provision and care of animals. Funding for this study was provided by the EPSRC Doctoral Training Centre in Regenerative Medicine.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.