
Abstract
Human embryonic stem cells and induced pluripotent stem cells have great potential in research and therapies. The current in vitro culture systems for human pluripotent stem cells (hPSCs) do not mimic the three-dimensional (3D) in vivo stem cell niche that transiently supports stem cell proliferation and is subject to changes which facilitate subsequent differentiation during development. Here, we demonstrate, for the first time, that a novel plant-derived nanofibrillar cellulose (NFC) hydrogel creates a flexible 3D environment for hPSC culture. The pluripotency of hPSCs cultured in the NFC hydrogel was maintained for 26 days as evidenced by the expression of OCT4, NANOG, and SSEA-4, in vitro embryoid body formation and in vivo teratoma formation. The use of a cellulose enzyme, cellulase, enables easy cell propagation in 3D culture as well as a shift between 3D and two-dimensional cultures. More importantly, the removal of the NFC hydrogel facilitates differentiation while retaining 3D cell organization. Thus, the NFC hydrogel represents a flexible, xeno-free 3D culture system that supports pluripotency and will be useful in hPSC-based drug research and regenerative medicine.
Introduction
H
To create a 3D stem cell niche, various cross-linked 3D hydrogels and scaffolds have been used to produce hPSC spheroids or aggregates [14 –17]. The cell spheroids and aggregates are formed in an environment with fixed physicochemical cues (eg, gelified alginate [15,17], polymerized hyaluronic acid [14], and polymer scaffold [16]) that are suitable only for stem cells and cannot be tuned during the culture to support differentiation processes in a variety of biomedical applications. To induce directed differentiation, the 3D stem cell-biomaterial organization should be destroyed, and the stem cells are transferred to a new system that is optimal for differentiation. This design is different from the natural stem cell niche in the blastocyst, which transiently supports stem cell proliferation and is subject to changes to facilitate subsequent differentiation during development. This concept has not yet been realized. A dynamic, flexible 3D in vitro culture system is still lacking.
Another approach to producing hPSCs aggregates or clusters is to use soluble factors instead of biomaterials in a suspension culture [18 –21]. However, karyotypic abnormalities were observed in some cell lines in the suspension culture [20]. Frequent dissociation of aggregates into single cells is needed, as the large aggregates develop necrosis in the center [21]. It is worth noting that the suspension culture does not provide a 3D environment for subsequent 3D differentiation.
We recently showed that a novel nanofibrillar cellulose (NFC) hydrogel promotes the functional 3D spheroid formation of human liver cells [22]. NFC is a plant-derived material with fiber diameter in nanometer range and fiber length in micrometer range. These fibrils are composed of aligned β-D-(1→4)glucopyranose polysaccharide chains [23]. NFC can be isolated from the cell walls of wood and plants. They form hydrogels with tuneable physical and chemical properties [24,25] and are shown to be non-cytotoxic [26]. Therefore, they have diverse pharmaceutical and biomedical applications [27 –29]. When used in cell culture, the NFC hydrogel is mixed with cells without any polymerization process.
In this study, we demonstrate, for the first time, that the NFC hydrogel creates a flexible 3D environment for proliferation and differentiation of hPSCs.
Materials and Methods
Generation of H9-GFP
H9-GFP cells (clone 24) were prepared by clonal selection of H9 cells (also known as WiCell WA09; WiCell) that were lentivirally transducted with rLV-EF1-GFP (Vectalys). Before infection, cells were disaggregated to single cells using accutase and plated in Corning non-adherent plates in hESC medium. After a 2-h transduction using a multiplicity of infection of 50, cells were washed, re-suspended in fresh medium, and seeded onto STO feeders. GFP-positive colonies arising from single cells were identified by fluorescence microscopy and isolated mechanically for passaging.
Cell culture on the standard Matrigel platform and in the NFC hydrogel
H9 (WA09) and WA07 [1] are hESC lines. iPS(IMR90)-4 [3] is a hiPSC line. WA07 and iPS(IMR90)-4 were purchased from WiCell. H9-GFP cell line was modified from H9 cell line that had been originally from WiCell. The cells were maintained on Matrigel-coated dishes in mTeSR1 medium (STEMCELL Technologies) at 37°C in a humid atmosphere with 5% CO2. Matrigel coating was prepared by incubating Matrigel (Matrigel™ basement membrane matrix growth factor reduced from BD Biosciences) dilution (0.5 mg per one 6-well plate) in tissue-culture plates at room temperature for 1 h as described in the WiCell stem cell protocol. The medium was renewed daily, and the cells were passaged at a ratio of 1:4 to 1:10 every 4–5 days using either 1 mg/mL dispase solution (STEMCELL Technologies) or Versene 1:5,000 (Invitrogen) for 7 min. The differentiated cells were removed by a pipette before splitting.
The colony density of hESCs and hiPSCs in 0.5 wt.% NFC hydrogel was five times higher than that of the culture in the Matrigel platform before passaging. 1.8 wt.% of NFC hydrogel stock solution (GrowDex™; UPM-Kymmene Corporation) was prepared from bleached birch pulp without any additional chemical modification. The NFC hydrogel is a mixture of cellulose and hemicellulose macromolecules and pure water. The hemicellulose fraction is mainly xylene, which generates slightly anionic surface charge (−2 mV) on the fibrillar structures. The overall carbohydrate composition is as follows: 72.8% Glucose, 25.6% Xylose, and 1.4% Mannose. The nanofibers are mainly composed of cellulose macromolecules, corresponding to the glucose fraction, and hemicellylose xylene is forming dangling chains on the fibrillar surfaces. 1.8 wt.% of NFC hydrogel stock solution was diluted in mTeSR1 medium and mixed with stem cell colonies. The same amount of mTeSR1 medium was added on top of cell-hydrogel mixture. The medium was renewed daily.
Subculture of hPSCs in the NFC hydrogel and recovery of 3D spheroids to 2D platforms
The cells cultured in the NFC hydrogel were passaged every 7–12 days to new NFC hydrogel or to 2D platforms. Cellulase (VTT) was used to subculture the 3D hPSCs and to recover spheroids from the hydrogel to 2D platforms. The optimal working temperature of cellulase is 45–50°C, which is, however, not suitable for human cells. The incubation time with cellulase should be extended to 24 h at 37°C. Before cellulase treatment, the old mTeSR1 medium was removed, and cellulase diluted in mTeSR1 medium was added and incubated with cell-hydrogel mixture at 37°C for 24 h.
After the enzymatic removal of NFC, hPSC spheroids were collected and treated with Versene 1:5,000 for 7 min at room temperature. The smaller colonies were generated by passing them through 1,000 μL pipette tip for ∼10 times. For 3D subculture, the smaller cell colonies were mixed with 0.5 wt.% NFC hydrogel as described earlier. To transfer colonies to 2D platforms, the smaller cell colonies were seeded on four different coatings: Matrigel, VN (R&D Systems), LN-511, and LN-521 (BioLamina) with a splitting ratio of 1:8 to 1:9. Matrigel coating was prepared as usual. LN-511 and LN-521 coatings were prepared by following the manufacturer's instructions. Briefly, 20 μg/mL LN-511 or 20 μg/mL LN-521 was incubated in tissue-culture plates at 37°C for 2 h and then at 4°C overnight. VN coating was prepared by incubating 5 μg/mL VN at 4°C overnight as previously described [6].
Mitochondrial metabolic activity
AlamarBlue® reagent (Invitrogen) was used to measure the mitochondrial metabolic activity of hPSCs before and after cellulase treatment in order to select a nontoxic enzyme concentration. Before and after treatment with various concentrations of cellulose, 2.5% AlamarBlue reagent was added into mTeSR1 medium and incubated with cells at 37°C for 24 h. The fluorescence of the AlamarBlue metabolite was measured at an excitation wavelength of 570 nm and an emission wavelength of 585 nm by a Varioskan Flash spectral scanning multimode reader 2.4.2 (Thermo Scientific).
NFC staining and live cell imaging using a confocal microscope
To evaluate the enzymatic removal of NFC, calcofluor white stain (Sigma FLUKA) was added into the culture to stain cellulose following the manufacturer's instructions. GFP fluorescence of the live H9-GFP cells and stained NFC were visualized under a Leica TCS SP5II HCS A confocal microscope using Argon 488 nm and UV 405 nm lasers, respectively, at 37°C with 5% CO2. Live/dead viability/cytotoxicity kit (Invitrogen) was used to assess cell viability in the spheroids.
Immunofluorescence and immunohistochemistry
hPSCs cultured either on different 2D platforms or in the NFC hydrogel were fixed in 3.7% paraformaldehyde for 10 min (2D) or 30 min (3D) at room temperature followed by permeabilization with 0.1% Triton X-100 or 0.5% saponin for 10 min (2D) or 30 min (3D). After blocking with 10% normal goat or donkey sera (Millipore), cells were incubated with anti-Oct-3/4 (Santa Cruz Biotechnology; sc-9081, 1:500), anti-SSEA-4 (Developmental Studies Hybridoma Bank; MC-813-70, 1:100), anti-β-tubulin isotype III (Sigma; T5076, 1:2,000), anti-AFP (Sigma; A8452, 1:500), anti-muscle actin (Dako; IS70030), or anti-HNF3B (Santa Cruz Biotechnology; sc-6554, 1:50) at 4°C overnight. Negative control samples incubated with control rabbit immunoglobulin G (IgG), mouse IgG, or goat IgG (Santa Cruz Biotechnology) were prepared in parallel. The secondary antibody, which was goat-anti-rabbit Alexa Fluor 594, goat-anti-mouse Alexa Fluor 594, or donkey-anti-goat Alexa Fluor 594 (Invitrogen) at a dilution of 1:400, was used at room temperature for 1 h (2D) or 6 h (3D). All washings using phosphate-buffered saline—0.2% Tween 20 were repeated thrice, 5 min each. After immunostaining, nuclei were stained with SYTOX Green (Invitrogen). Cells were then mounted with VECTASHIELD mounting medium (Vector Laboratories). The staining was viewed under a Leica TCS SP5II HCS A confocal microscope using Argon 488 nm laser for GFP and SYTOX Green and DPSS 561 nm laser for Alexa Fluor 594. The confocal images were analyzed with Imaris 7.4 software (Bitplane AG).
In some experiments, hPSC spheroids and embryoid bodies (EBs) were fixed in 3.7% paraformaldehyde and embedded in HistoGel (Thermo Scientific). Subsequently, the standard paraffin embedding and sectioning were performed at the Finnish Center for Laboratory Animal Pathology. Five-micrometer-thick sections were used in immunohistochemistry.
RNA extraction and real-time quantitative reverse transcription–polymerase chain reaction
Total RNA was extracted using RNeasy Mini (RNA from cultured cells) and RNeasy Midi kit (RNA from teratoma) (Qiagen) following the manufacturer's instructions. RNA samples were quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). All the RNA samples were converted into cDNA at the same experiment to ensure the same reverse transcription efficiency. The cDNA synthesis was performed by using a High-Capacity RNA-to-cDNA kit (Applied Biosystems). All the cDNA samples were analyzed in duplicate using a Fast SYBR Green Master Mix (Applied Biosystems) on a StepOnePlus Real-Time PCR System (Applied Biosystems). For each gene, a standard curve was generated, and amplification efficiency was taken into account in calculations. Polymerase chain reaction (PCR) product quality was monitored using post-PCR melt curve analysis. All primers were synthesized by Oligomer Oy. The housekeeping gene RPLP0 was used as an endogenous control. The PCR cycling conditions were as follows: 40 cycles of 3 s at 95°C and 30 s annealing/extension at 60°C. The primer sequences were shown in Table 1.
In vitro differentiation via EB formation
iPS(IMR90)-4 cells were cultured in 0.5% NFC hydrogel for 8 days and then treated with cellulase for 24 h. The EB medium consisting of Iscove's modified Dulbecco's medium (Invitrogen) supplemented with 15% HyClone-Defined fetal bovine serum (Thermo Scientific) was used in the EB formation. To form EBs, two methods were used. In a direct method, the cell spheroids that recovered from the hydrogel were directly cultured in suspension in the EB medium in Nunc HydroCell surface 3.5 cm dishes (NUNC) for 4 weeks. In an indirect method, cells were first recovered from the hydrogel and then cultured in mTeSR1 medium in Matrigel-coated dishes for 4 days. The 2D cell colonies were then used to form EBs in suspension in the EB medium for 4 weeks. To form EBs from 2D culture, the WiCell protocol was followed. In brief, cell colonies were detached from Matrigel-coated wells by 1 mg/mL dispase for 17 min at 37°C. Small colonies and debris were removed by a 100 μm cell strainer (BD Biosciences). The remaining colonies were cultured in the EB medium in Nunc HydroCell surface 3.5 cm dishes for 4 weeks.
At every week, EBs were collected and dissociated using Versene 1:5,000. The resulting cells were seeded on Lab-Tek eight-well chamber slides (NUNC) that were coated with ES-qualified 0.1% gelatin (Millipore) in the EB medium for an additional week of culture. The cells were then fixed in 3.7% paraformaldehyde for 10 min at room temperature and detected by immunofluorescence. In addition, at every week, EBs were collected, fixed in 3.7% paraformaldehyde for 30 min at room temperature, and embedded in HistoGel (Thermo Scientific). Immunohistochemistry was performed as described in the earlier section.
Teratoma formation
After a 26-day culture of WA07 cells in 0.5 wt.% NFC hydrogel, the cell spheroids were harvested after cellulase treatment, collected into tubes, and pelleted by centrifugation. Teratoma assays were performed by injecting the spheroids into the testis of two nude NMRI mice at the Biomedicum Helsinki Stem Cell Center. The tumors were harvested at 6 weeks after the injection, fixed in 4% paraformaldehyde, and processed for paraffin embedding. Hematoxylin- and eosin-stained sections (5 μm) were morphologically analyzed for the presence of derivatives of all germ layers. Parts of the fresh tumors were stored in RNAlater RNA stabilization reagent (Qiagen) for subsequent RNA isolation and real-time quantitative reverse transcription–polymerase chain reaction (RT-PCR).
Karyotyping
For karyotyping analyses, WA07 and iPS(IMR90)-4 cell spheroids were recovered from the hydrogel and seeded to Matrigel-coated dishes following the procedure mentioned earlier. Chromosomal G-band analyses were performed at the United Medix laboratories Ltd.
Statistical analysis
Statistical significance was determined by one-way analysis of variance followed by Holm–Sidak post-test (SigmaPlot). Differences of P>0.05 were not considered significant, and P<0.01 (**) and P<0.001 (***) were considered significant (Figs. 2 and 4).
Results
Formation of 3D hPSC spheroids in the NFC hydrogel
In this study, we explored the possibility of creating 3D hPSC spheroids in a flexible environment that enables further differentiation without disrupting 3D stem cell organization. We cultured hPSCs in a novel NFC hydrogel and generated 3D spheroids. The NFC hydrogel concentration affects 3D spheroid formation. hPSCs failed to form spheroids in 1 wt.% NFC hydrogel (Supplementary Fig. S1; Supplementary Data are available online at
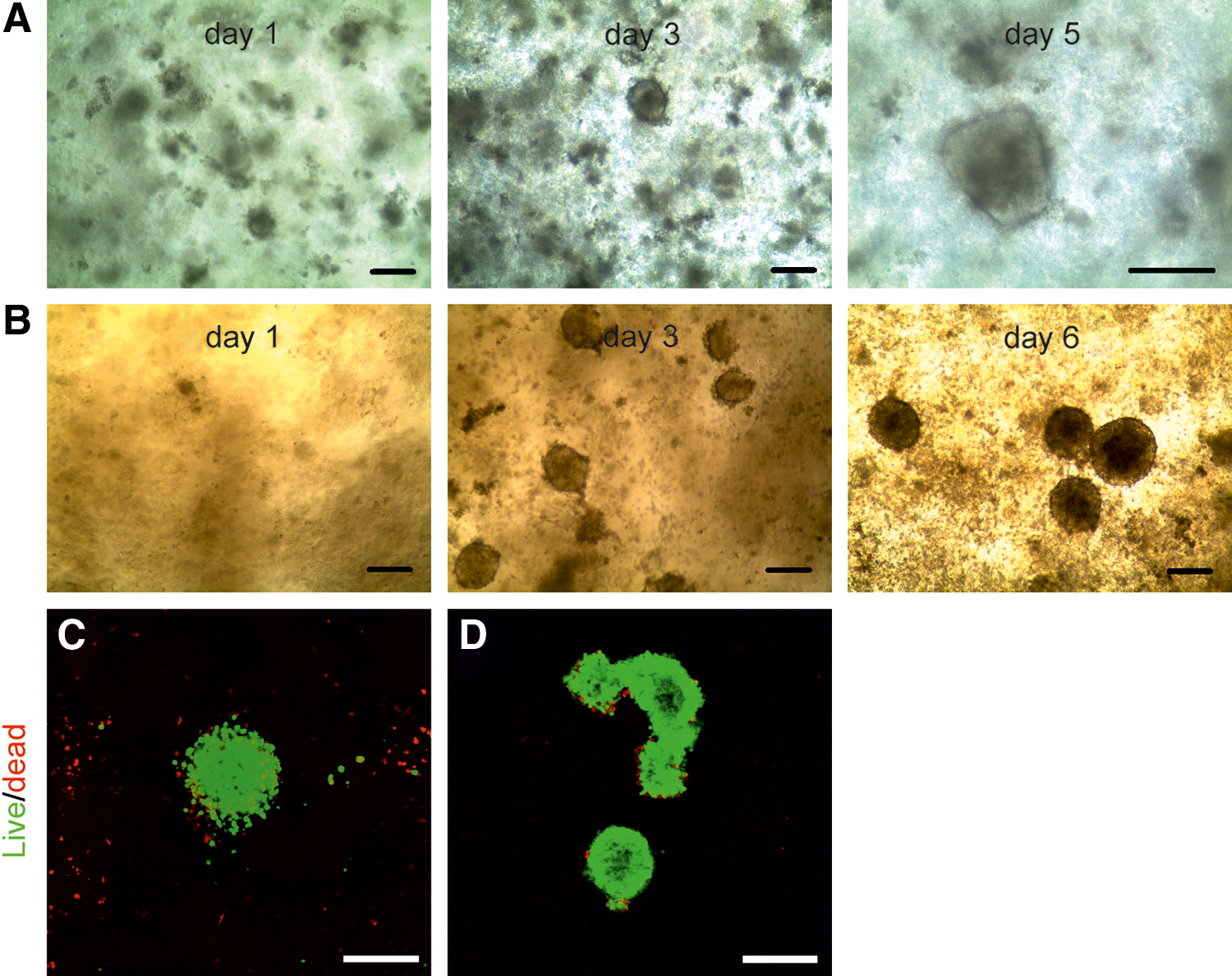
Human embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) form three-dimensional (3D) spheroids in the nanofibrillar cellulose (NFC) hydrogel.
Enzymatic removal of the NFC hydrogel
We made use of cellulase to subculture 3D hPSCs and to recover spheroids from the hydrogel to 2D platforms. Cellulase is an enzyme that specifically degrades cellulose without affecting animal cells, because animal cells do not contain cellulose [30,31]. The degraded products of the NFC are nontoxic sugars. Before using cellulase in the 3D culture, we optimized cellulase concentration in a standard Matrigel platform for each hPSC line. AlamarBlue assay was used to assess cell viability and the possible toxicity of cellulase. Figure 2A and B show that cellulase at concentrations no more than 300 μg/mg cellulose had no statistically significant effect on the viability and growth of WA07, iPS(IMR90)-4, and H9-GFP cells. The removal of cellulose was visualized by calcofluor white stain, which is a cellulose-binding fluorochrome. The use of H9-GFP cells enabled us to monitor the spheroid structure after the enzymatic removal of the NFC hydrogel. The 3D spheroids stayed intact after the treatment with cellulase at 50, 200, and 500 μg/mg cellulose, and cellulase below 200 μg/mg cellulose was insufficient to remove NFC (Fig. 2C). OCT4 was immunodetected in H9-GFP cell spheroids after the enzymatic removal of the NFC hydrogel (Fig. 2D).
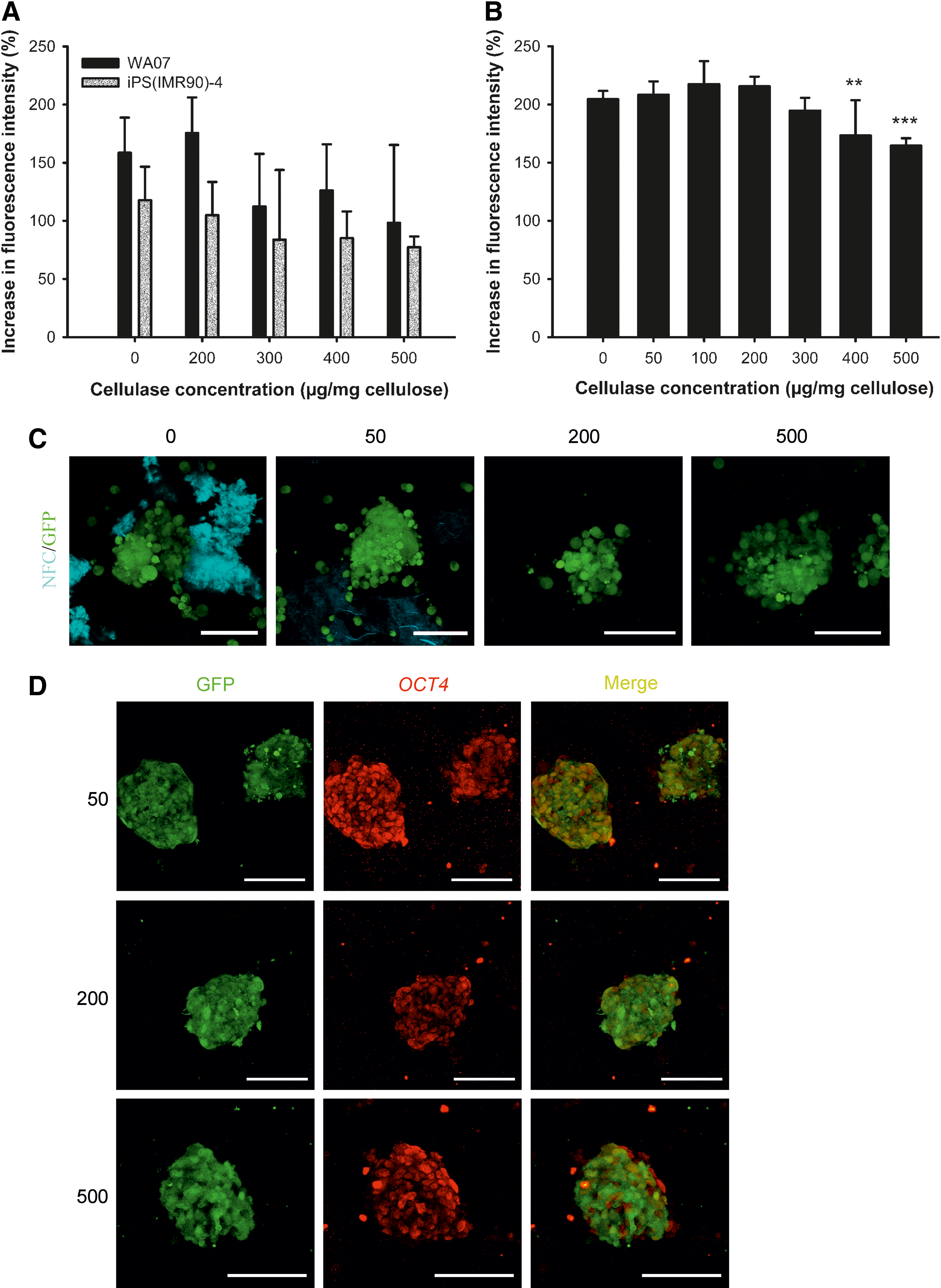
Enzymatic removal of the NFC hydrogel.
Pluripotency of hPSCs cultured in the NFC hydrogel
To determine whether hPSCs cultured in the NFC hydrogel are pluripotent, we performed extensive analyses, including pluripotency marker expression, in vitro EB-mediated differentiation, and teratoma assay. WA07 (Fig. 3A–D) and iPS(IMR90)-4 (Fig. 3E) cells in 3D spheroids during 26-day culture expressed the pluripotency marker OCT4 shown by immunofluorescence. In contrast, iPS(IMR90)-4 (Supplementary Fig. S2A) and WA07 (Supplementary Fig. S2B, C) cells did not express the differentiation markers β-tubulin type III, HNF3B, and muscle actin. Real-time quantitative RT-PCR shows that during a 26-day culture in the hydrogel, WA07 cells in the spheroids expressed a similar amount of NANOG mRNA as cells cultured on the standard Matrigel platform (M ctrl) (Fig. 4B). The expression of OCT4 mRNA in WA07 spheroids was maintained at day 7 but dropped by ∼50% at day 26 (P<0.001, Fig. 4A). A possible explanation is that the manual removal of spontaneous differentiation was not performed in the 3D culture, whereas it is a routine procedure before every subculture on the standard Matrigel platform.
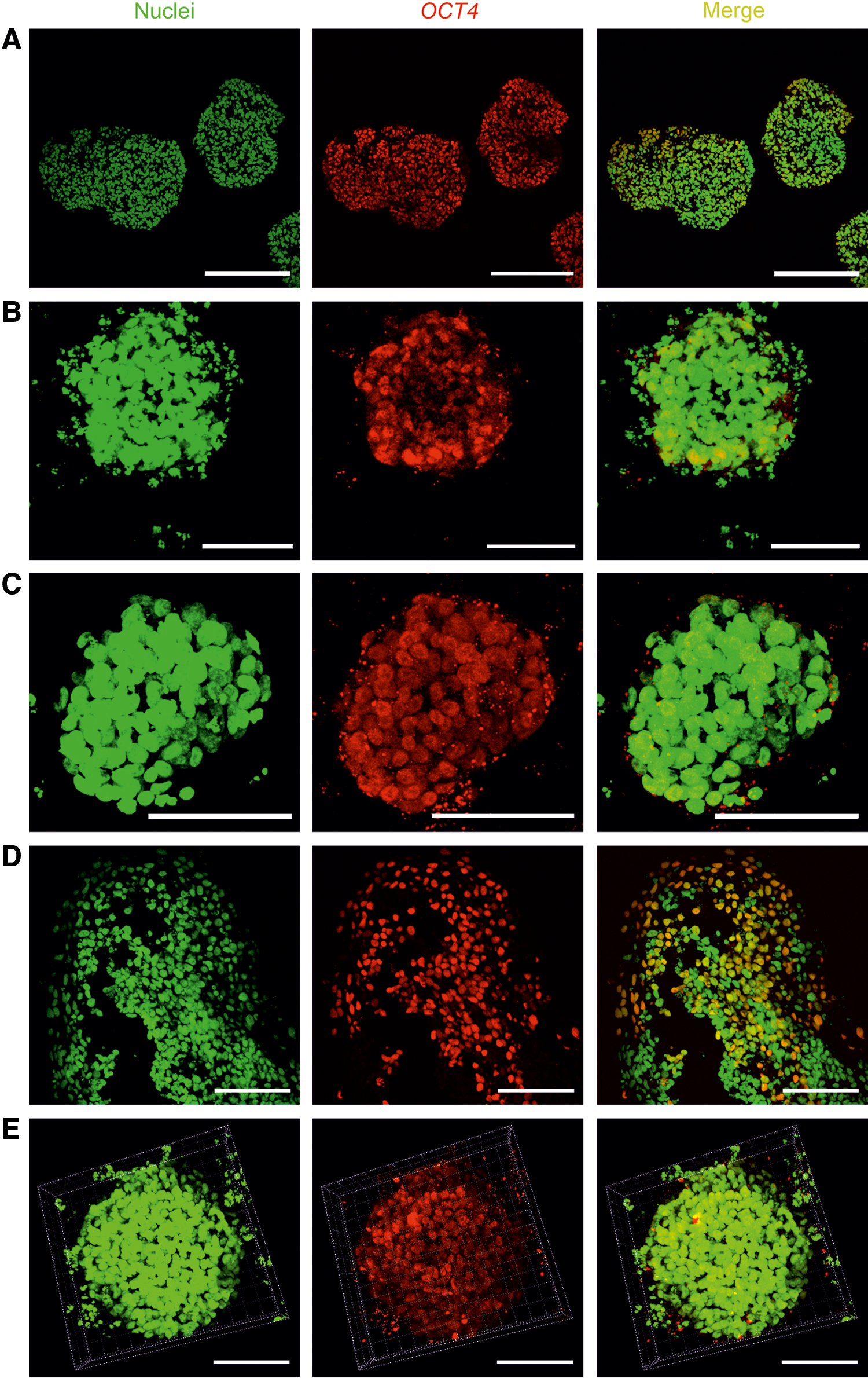
OCT4 expression in the 3D human pluripotent stem cell (hPSC) spheroids. The pluripotency marker OCT4 is expressed in WA07 cells that are cultured in 0.5% NFC hydrogel for 7 days [
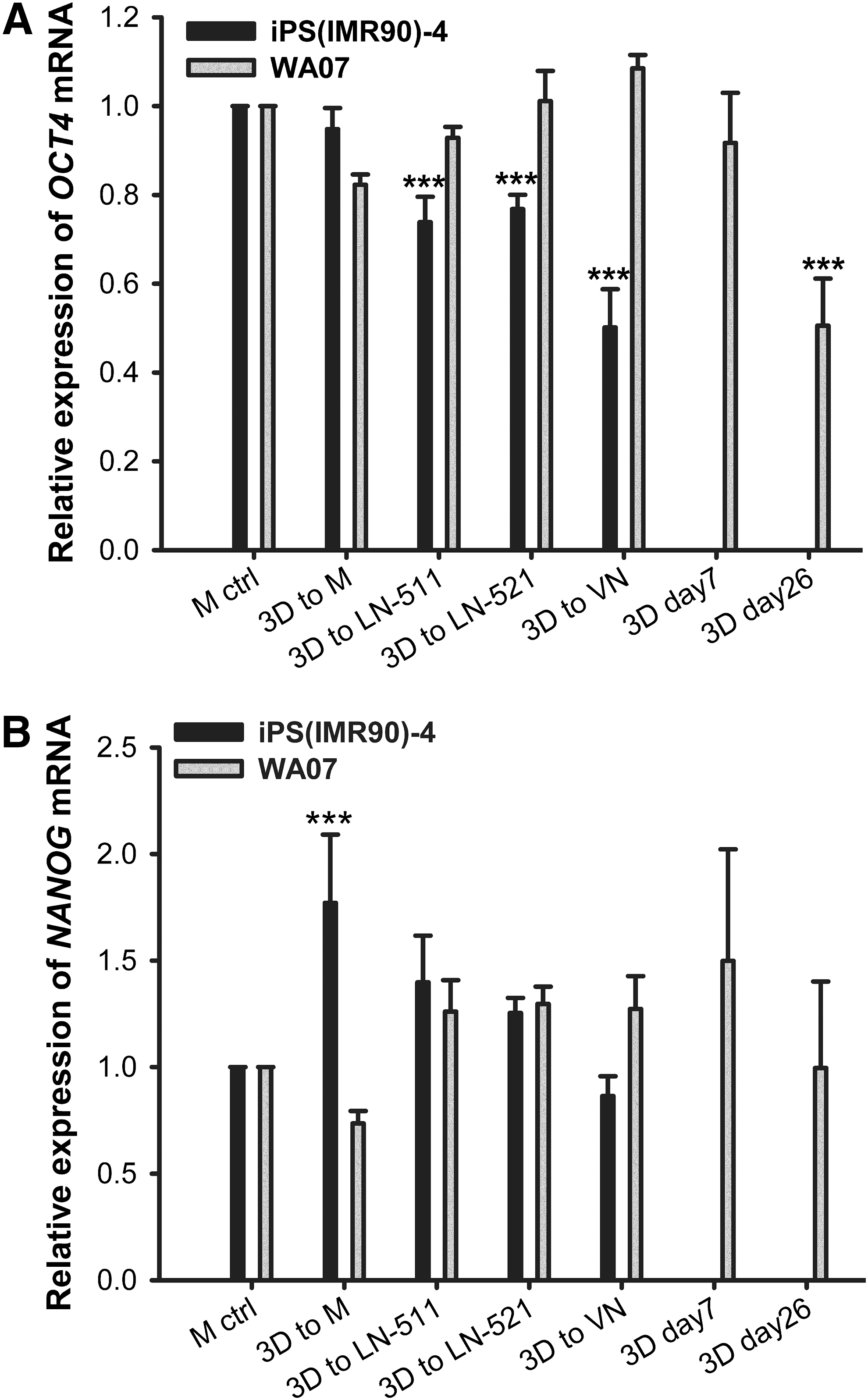
Gene expression in hPSCs cultured in 3D and two-dimensional (2D) platforms. Real-time quantitative reverse transcription–polymerase chain reaction (RT-PCR) analyses of OCT4
The 3D culture can be easily shifted to various 2D platforms. We used four 2D platforms, namely Matrigel coating, LN-511 coating, LN-521 coating, and VN coating. By using 300 μg cellulase/mg cellulose at 37°C for 24 h, we were able to recover the intact spheroids. WA07 cells from the recovered spheroids possessed the characteristics of undifferentiated hESCs when they were plated to the Matrigel, LN-511, LN-521, and VN coatings (Fig. 5A–D). They also expressed OCT4 and SSEA-4 proteins as shown by immunofluorescence (Fig. 5A–D). We obtained the same results using iPS(IMR90)-4 (Fig. 6) and H9-GFP (Supplementary Fig. S3) cells. The differentiation markers β-tubulin type III, HNF3B, and muscle actin were not detected in WA07 cells (Supplementary Fig. S4) and iPS(IMR90)-4 cells (Supplementary Fig. S5). After being cultured in the NFC hydrogel, WA07 cells maintained the expression of OCT4 and NANOG mRNA on the Matrigel, LN-511, LN-521, and VN coatings (Fig. 4). iPS(IMR90)-4 cells maintained the expression of NANOG mRNA (Fig. 4B) but showed a reduction in OCT4 mRNA expression when cultured in LN-511, LN-521, and VN (Fig. 4A). When comparing the four 2D platforms, we noticed that Matrigel coating was the best to maintain high levels of NANOG and OCT4 mRNA expression (Fig. 4).
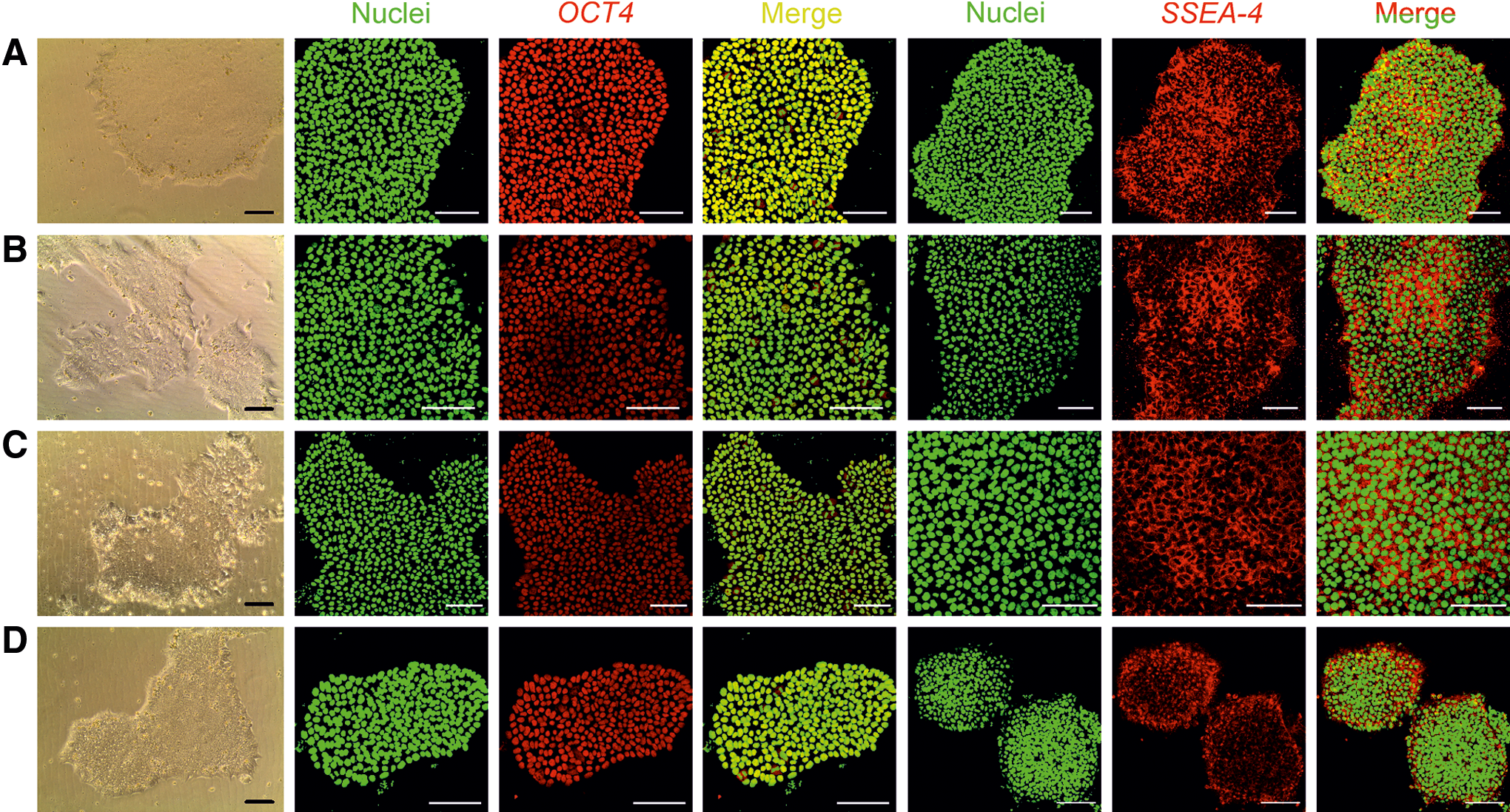
Transfer of WA07 3D spheroids to 2D platforms.
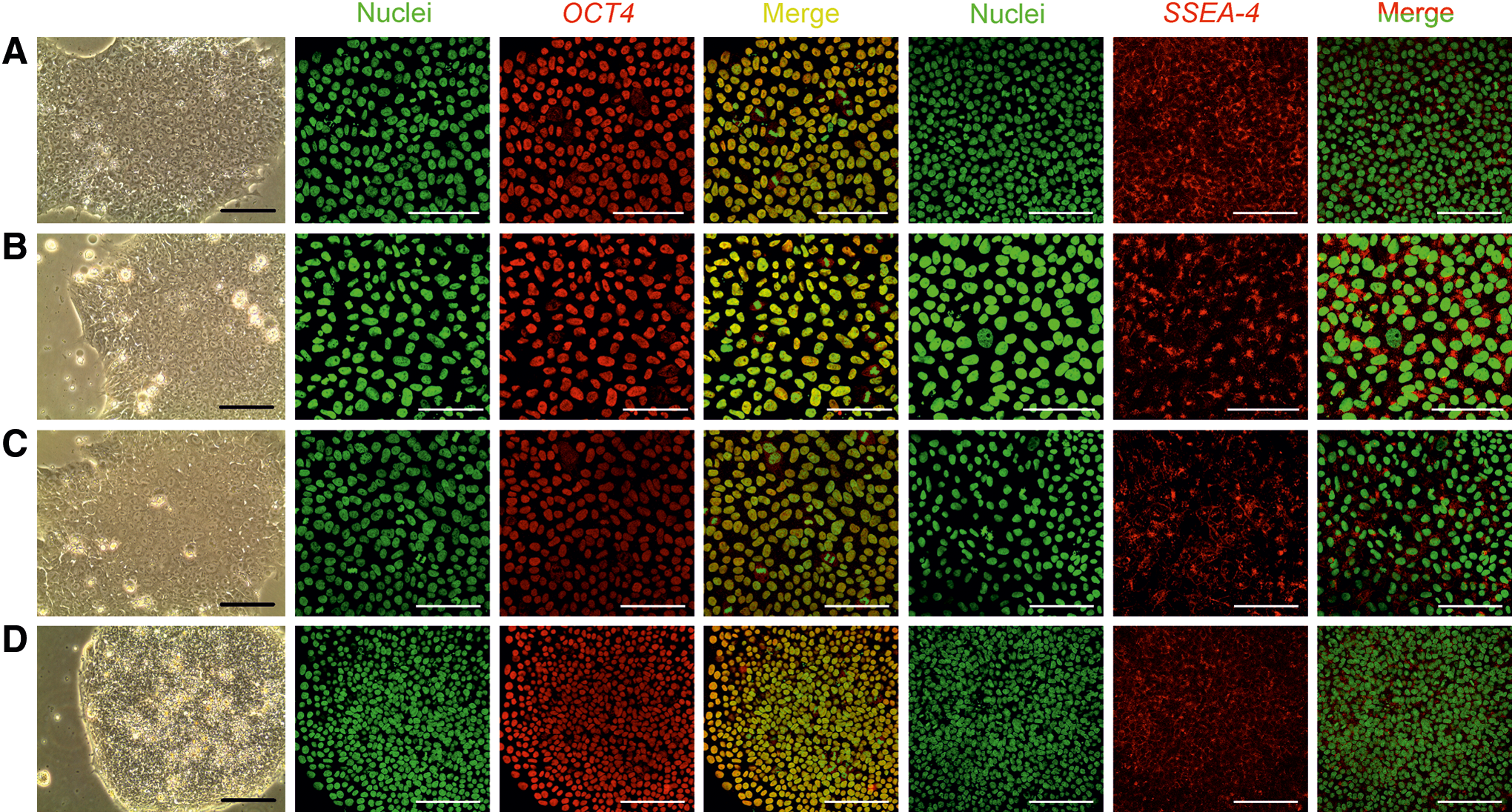
Transfer of iPS(IMR90)-4 3D spheroids to 2D platforms. iPS(IMR90)-4 cells were first cultured in 0.5% NFC hydrogel for 12 days and then transferred to 2D platforms after a 24-h-treatment with 300 μg cellulase/mg cellulose at 37°C. The cells exhibited typical hPSC morphology and expressed the pluripotency markers OCT4 and SSEA-4 on Matrigel-coated dishes
We demonstrate that after being cultured in the NFC hydrogel, iPS(IMR90)-4 cells were able to differentiate into three germ layers as shown by immunostaining of β-tubulin type III, muscle actin, and AFP (Fig. 7A) and real-time quantitative RT-PCR analyses of PAX6, CDX2, and BRACHYURY (Fig. 7B). The EBs derived from iPS(IMR90)-4 cells exhibited tightly packed morphology (dark EBs) during the first week of EB culture (Supplementary Fig. S6A). The other types of EBs, such as bright EBs and cystic EBs, appeared after a 1-week culture (Supplementary Fig. S6B). iPS(IMR90)-4 cells also differentiated into pigmented cells (Supplementary Fig. S6C) and developed beating “hearts” in the third week of EB culture (Supplementary Video S1). The EB-mediated differentiation potency of iPS(IMR90)-4 cells did not differ between EBs formed directly from 3D spheroids and those formed indirectly after a 3D to 2D shift. Immunofluorescence in the older EBs shows strong staining of muscle actin, HNF3B, and β-tubulin type III (Supplementary Fig. S6D–F).
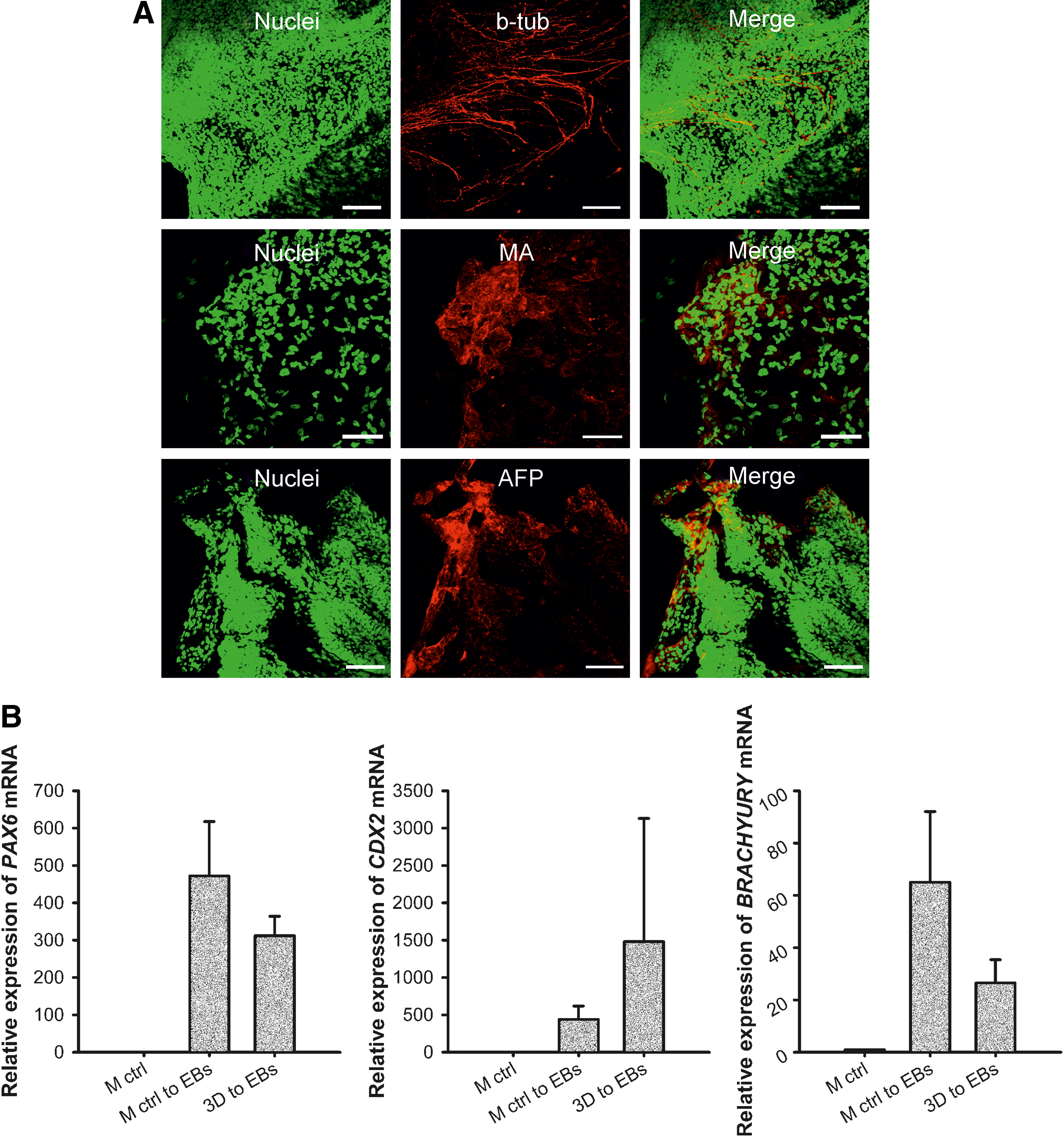
In vitro differentiation of iPS(IMR90)-4 cells.
The teratoma formation from WA07 cells after a 26-day culture in the NFC hydrogel provides more solid evidence, showing that the 3D culture of hPSCs in the hydrogel does not alter the pluripotency of hPSCs. We observed the three germ layer derivatives in hematoxylin and eosin staining of 6-week-old teratoma sections (Fig. 8A–J). Immunofluorescence in the teratoma sections shows strong staining of β-tubulin type III and muscle actin (Supplementary Fig. S7). The mRNA expression of PAX6, CDX2, and BRACHYURY was dramatically increased in teratoma samples compared with cells on the standard Matrigel platform (M ctrl) (Fig. 8K).
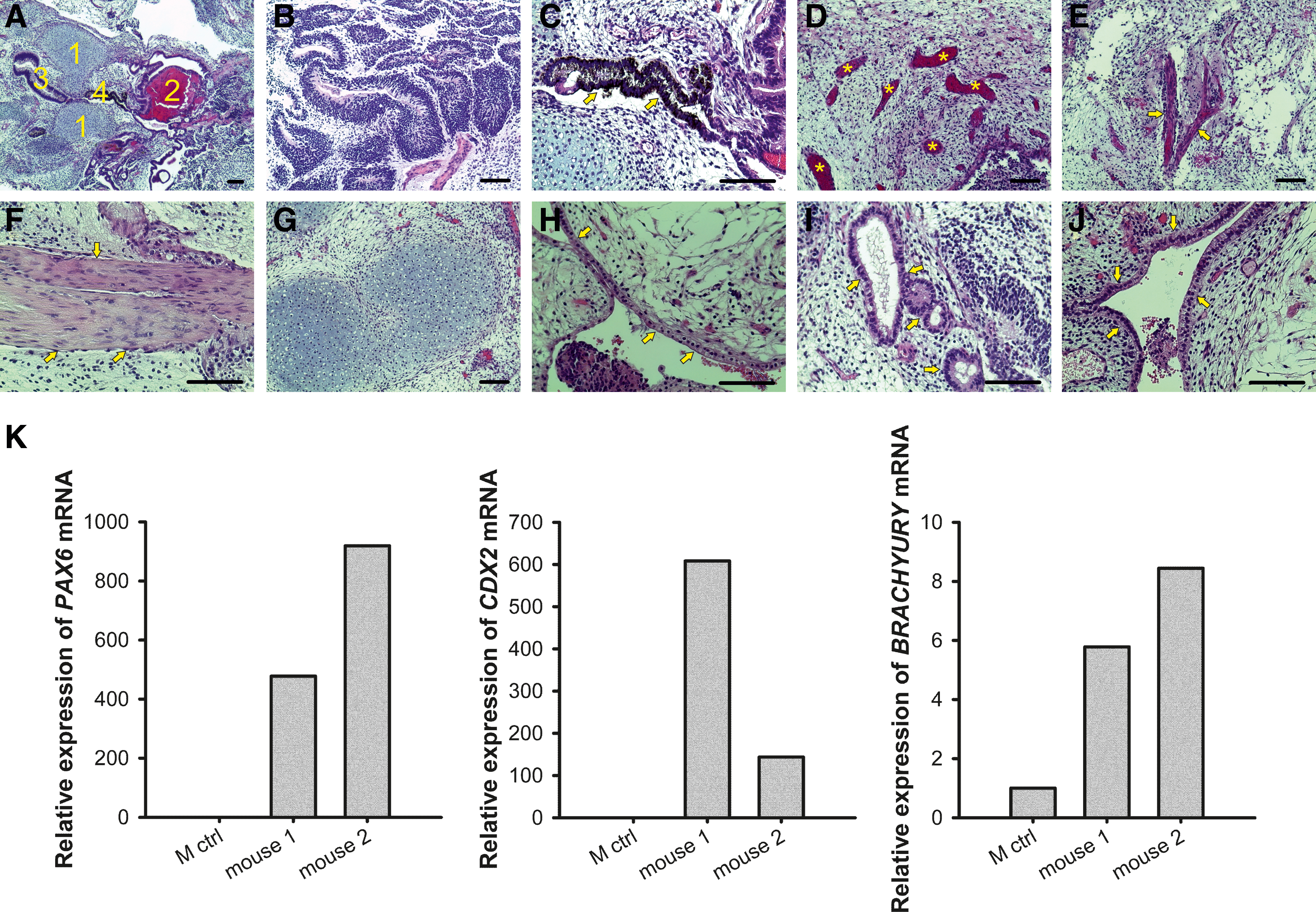
In vivo differentiation of WA07 cells.
Chromosomal G-band analyses show that after being cultured in the NFC hydrogel, WA07 and iPS(IMR90)-4 cells had normal karyotypes (Fig. 9).
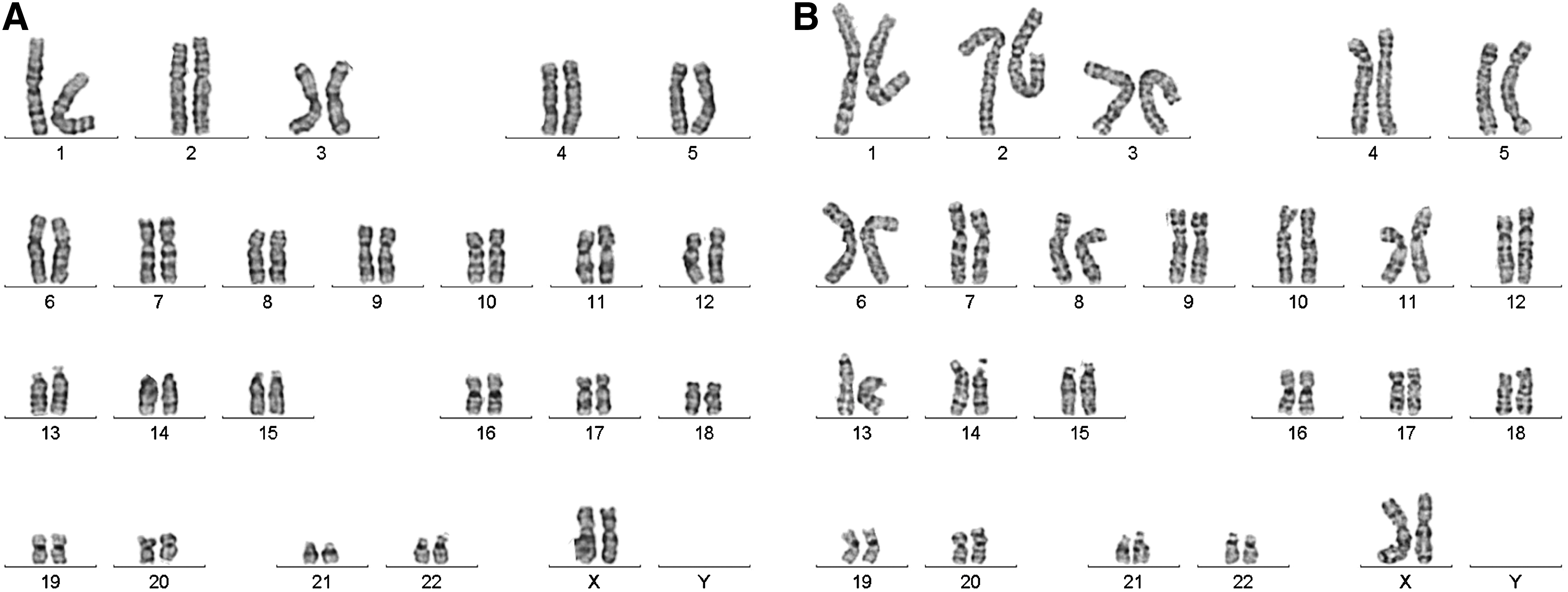
The karyotypes of human ESCs and iPSCs. WA07 and iPS(IMR90)-4 cells were first cultured in 0.5% NFC hydrogel and then transferred to Matrigel-coated dishes after a 24-h treatment with 300 μg cellulase/mg cellulose at 37°C. G-banding chromosome analysis showed normal chromosomes in WA07, p41
Discussion
The natural stem cell niche is a dynamic 3D environment that transiently supports stem cell proliferation and is subject to changes which facilitate subsequent differentiation during development. Here, we propose a new concept of hPSC culture, which is a flexible, xeno-free 3D in vitro culture system. This system is based on a novel plant-derived NFC hydrogel. Unlike most polysaccharide-based hydrogels that require a separate cross-linking step to form the hydrogel networks [32], the NFC forms colloidal dispersions and hydrogels in aqueous medium [33]. Thus, the hydrogels generated by cross-linking have fixed physical properties, whereas the physical property of the NFC hydrogel is dependent on its concentration, not on the degree of cross-linking. In addition, enzymatic removal of the NFC hydrogel does not affect the cells. All these features make the NFC hydrogel a flexible 3D environment.
We demonstrate that the pluripotency of hPSCs is maintained in the NFC hydrogel for 26 days. The NFC hydrogel promotes cell–cell associations into 3D spheroids with the expression of the pluripotency markers. This 3D culture can be easily shifted to 2D platforms, enabling various downstream applications. Since enzymatic removal of the hydrogel does not affect 3D cell organization, the established 3D hPSC spheroids are ready for directed differentiation into various types of cells and even 3D tissues, as shown in EB formation by the direct method.
We previously showed that the mechanical property (eg, viscoelasticity) of the NFC hydrogel is controllable and dependent on the NFC concentration [22]. The lower the NFC concentration is, the lower the viscoelasticity of the hydrogel is. In this study, we show that hPSCs form 3D spheroids in 0.5 wt.% NFC hydrogel, but not in 1 wt.% NFC hydrogel. This indicates that the lower concentration of the NFC hydrogel with less viscoelasticity is preferable for 3D hPSC spheroid formation. The mechanical properties of the human inner cell mass at the blastocyst stage are not known. An early study showed that soft substrates with elastic Young's modulus of 0.6 kPa promote self-renewal and pluripotency of mouse ESCs [34]. Since no cross-linking is involved in the formation of the NFC hydrogel, the mechanical property of the hydrogel can be adjusted by its concentration for subsequent differentiation steps.
To our knowledge, this is the first study using LN-521 coating in hPSC culture and also the first comparison between Matrigel coating, LN-511 coating, LN-521 coating, and VN coating in hPSC culture. It is interesting to notice that the expression of OCT4 mRNA was not well maintained in hiPSCs cultured on LN-511, LN-521, and VN coatings (Fig. 4A). VN had not been demonstrated in hiPSC culture in the previous study [6]. The expression of OCT4 mRNA in hiPSCs cultured on LN-511 coating was not demonstrated in the earlier study [7]. Nonetheless, Matrigel coating seems to be the best to maintain high levels of NANOG and OCT4 mRNA expression, although there is a cell type-dependent variation.
Earlier studies show that the pluripotency of hPSCs requires well-established cell–cell contact, which is mainly via E-cadherin [35 –37]. On the other hand, cell–matrix interaction seems not to be a prerequisite as evidenced by the successful use of poor cell-adhesive hyaluronic acid hydrogel [14], alginate [15,17], and the NFC hydrogel of the current study in hPSC culture. The non-adhesive feature of the NFC hydrogel is the driving force for the formation of 3D spheroids. Contrary to 3D culture, cell–matrix interaction mediated by integrins seems to be important for hPSC survival and self-renewal in 2D cultures [4,6,38,39].
Conclusions
This study provides a new concept of using a flexible, xeno-free 3D culture system for pluripotency of hPSCs and opens up a new avenue for better recapitulation of the natural ESC environment and embryonic development, which 2D cultures and conventional cross-linked 3D hydrogels and scaffolds could not attain.
Footnotes
Acknowledgments
The authors would like to thank the Academy of Finland and UPM-Kymmene Corporation for the financial support received. This research belongs to the “Biocenter Finland platform” project. The authors also want to thank Leena Pietilä and Timo Oksanen for reagent purchasing and Erja Piitulainen for laboratory logistics.
Author Contributions
Y.-R.L. conceived the study and experimental design, performed and analyzed experiments, and wrote the article. L.K., T.K., and J.N. performed experiments. L.A.N. and D.B. provided H9-GFP cells. A.U. provided the initial funding of the project. M.Y. provided conceptual support and funded the project. All authors read and approved the final manuscript.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.