
Abstract
Reprogramming of somatic cells toward pluripotency involves extensive chromatin reorganization and changes in gene expression. Polycomb group (PcG) proteins are key regulators of chromatin structure, cell identity, and development. In this study, we investigated the impact of Ezh2, a core subunit of Polycomb repressive complex 2 (PRC2), on the generation of induced pluripotent stem (iPS) cells. We found that Ezh2 expression is induced during iPS cell generation and iPS cells contain high levels of Ezh2 mRNA and protein. Importantly, shRNA knockdown of Ezh2 during reprogramming severely impairs iPS cell generation. Mechanistically, Ezh2 acts during reprogramming at least in part through repressing the Ink4a/Arf locus, which represents a major roadblock for iPS cell generation. Interestingly, knockdown of Ezh2 in established pluripotent cells leaves pluripotency and self-renewal of embryonic stem cells and iPS cells unaffected. Altogether, our results demonstrate that Ezh2 is critical for efficient iPS cell generation, whereas it is dispensable for maintaining the reprogrammed iPS cell state.
Introduction
P
PcG proteins are also required for establishing ES cell lines and for reprogramming somatic cells toward pluripotency. For example, blastocysts deficient for the PRC2 component Ezh2 failed to yield ES cells or produced ES cells at very low frequency [7,8]. ES cells lacking the PRC2 components, Ezh2, Eed, and Suz12, were deficient in cell fusion-induced reprogramming of somatic cells toward pluripotency [9]. In somatic cell nuclear transfer (SCNT) experiments, the inner cell mass of cloned embryos showed low H3K27me3 modification compared to fertilized embryos and thus differentiation-related genes were expressed [10]. Furthermore, the low levels of H3K27me3 in SCNT embryos correlate with low Ezh2 expression in such cloned embryos. All these studies support the notion that PcG proteins contribute to establish pluripotency.
Induced pluripotent stem (iPS) cells are generated from somatic cells by transduction of specific reprogramming transcription factors [11]. iPS cells hold great potential in disease modeling, drug discovery, and cell-based therapies [12,13]. iPS cell generation is regulated by a series of complex processes that are increasingly being better understood [14 –19]. Extensive epigenetic reorganization occurs during reprogramming, and recent studies indicate that activities of epigenetic modifiers play an important function in reprogramming, and thus, the role of PcG proteins in iPS cell generation is now beginning to be studied in detail [15,19 –24].
Here, we investigated the impact of the PcG protein Ezh2 on iPS cell generation. We studied the influence of Ezh2 overexpression and knockdown on iPS cell generation. We show that Ezh2 is critical for efficient iPS cell generation and acts—at least in part—through repressing the cell cycle regulator Ink4a/Arf.
Materials and Methods
Cells and cell culture
Mouse embryonic fibroblasts (MEF) were isolated from C57BL/6 mice or Oct4-eGFP transgenic OG2 mice [25]. MEF and 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen), containing 10% FCS, 2 mM
ES cells (HM1) and iPS cells were cultured, as described previously [26,27]. Briefly, cells were maintained on inactivated MEF feeder layer from C57BL/6 mice with ES cell medium. The ES cell medium was DMEM (high glucose), supplemented with 15% heat-inactivated fetal calf serum (FCS; Lonza), 25 mM HEPES, and 1,000 U recombinant leukemia inhibitory factor, 2 mM
Plasmids, virus preparation, and iPS cell generation
pMX retrovirus vectors with reprogramming factors, Oct4, Sox2, Klf4, and c-Myc, were used for iPS cell generation [11,28]. MSCV retrovirus vector containing mouse Ezh2 cDNA was obtained from Addgene (
For virus preparation, 293T cells were transfected with vector constructs and packaging plasmids (pVPack-GP and pVPack-Eco; Stratagene). shRNA lentivirus vectors targeting Ezh2 were transfected into 293T cells with the packaging plasmids pCMVΔR8.74 and pVSV-G, as described previously [26]. Transfections were with the calcium phosphate coprecipitation method. Forty-eight hours after transfection, virus supernatant was harvested and concentrated by precipitation with chondroitin 6-sulfate (CSC) and polybrene (both Sigma) and centrifuged [33].
For iPS cell generation, 5×104 MEF of C57BL/6 mice or Oct4-eGFP transgenic OG2 mice were seeded per well of six-well plate the day before infection and exposed to concentrated pMX viruses with Oct4, Sox2, Klf4, and c-Myc for 24 h and were cultured in the ES cell medium. For OG2 MEF as targets, GFP-positive (GFP+) colonies appeared around day 14 after virus transduction. GFP+ iPS cells were picked at day 18 and transferred into 96-well format. For C57BL/6 MEF, kinetics of iPS cell generation was similar to OG2 MEF iPS cells, and emerging iPS cells were identified by morphology and picked 2–3 weeks after infection. Single cell suspensions were obtained by trypsin treatment, and cells were cultured in 24-well plates and further in 6-well plates on MEF feeder in the ES cell medium.
Alkaline phosphatase staining
Emerging iPS cells were subjected to alkaline phosphatase (AP) staining by using the Alkaline Phosphatase Staining Kit (Stemgent) according to the manufacturer's protocol. Stained cells were photographed, and AP+ colonies were scored.
Cell cycle analysis and β-galactosidase staining
For cell cycle analysis, cells were fixed, incubated with 7-aminoactinomycin D (7-AAD) (10 μg/mL; Molecular Probes, Invitrogen), as described previously [34] and submitted to the flow cytometry analysis (FACSCanto device; BD Biosciences). Data were analyzed with FlowJo software (Tree Star).
MEF were measured for the β-galactosidase (β-gal) activity using the β-gal staining kit (Cell Signaling Technology).
DNA and RNA isolation and polymerase chain reaction
For DNA isolation, the NucleoSpin Tissue Kit (Macherey-Nagel) was used and 50 ng total DNA was subjected to quantitative polymerase chain reaction (qPCR) with SYBR Green PCR master mix and eGFP-specific primers (see Supplementary Table S1;Supplementary Data are available online at
Total RNA was isolated with the RNeasy Mini Kit (Qiagen). One microgram of total RNA was used for cDNA preparation with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). cDNA was then used for PCR amplification by Taq DNA polymerase (Fermentas Life Sciences). PCR fragments were separated on 2% agarose gels, and images were recorded with the Gel-Doc system (BioRad).
Quantitative real time-PCR (qRT-PCR) was carried out with the Applied Biosystems 7300 Real-Time PCR system (Applied Biosystems). Reactions were performed with 50 ng cDNA, SYBR Green PCR master mix, and primers (see Supplementary Table S1). The calculated threshold cycle (CT) value for each sample was normalized against the corresponding β-actin CT value.
Western blotting and immunofluorescence
Cells were lysed in 2% SDS with 5 mM EDTA. Twenty micrograms of protein lysate was subjected to SDS-PAGE (12% SDS-polyacrylamide gels) and western blotting. Membranes were blocked overnight with 2% nonfat milk in PBS at 4°C and incubated with primary antibodies (mouse anti-Ezh2, AC22, 1:1,000, Cell Signaling Technology; rabbit polyclonal anti-H3K27me3, 1:100, Millipore; mouse anti-Oct3/4, C-10, 1:200, Santa Cruz Biotechnology; mouse anti-actin antibody, clone AC-74, 1:5,000, Sigma) at room temperature for 2 h. Peroxidase-conjugated secondary antibodies were incubated for 1 h at room temperature and subjected to chemiluminescence (SuperSignal®; Thermo Scientific).
For immunofluorescence staining, ES cells and iPS cells were grown on gelatin-coated chamber slides, fixed with 1% formaldehyde, and permeabilized with PBS buffer containing 0.5% BSA and 0.1% Triton X-100. Cells on coverslips were washed two times with 0.5% BSA in PBS buffer and then incubated with primary antibodies (mouse anti-Ezh2, clone AC22, 1:100, Cell Signaling Technology; mouse anti-SSEA-1, clone 480, 1:50, Santa Cruz Biotechnology; and mouse anti-Oct3/4, clone C-10, 1:100, Santa Cruz Biotechnology). Secondary antibody was anti-mouse Alexafluor647 (Invitrogen), and DAPI was used to stain nuclei. Coverslips were mounted onto glass slide with the mounting medium, and images were acquired under bright and fluorescent fields with an Axiovert 200 microscope (Carl Zeiss).
Teratoma formation assay
NOD/SCID mice were maintained under specific pathogen-free conditions in the central animal facility of the RWTH Aachen University Hospital. All animal experiments were approved by local authorities in compliance with the German animal protection law.
For teratoma formation assay, 2×106 iPS cells were injected into the rear thigh of NOD/SCID mice. HM1 ES cells were used as control. After 1 month, mice were sacrificed, teratomas excised, fixed in 4% paraformaldehyde, embedded in paraffin, sectioned, and analyzed after hematoxylin and eosin (HE) staining.
Statistics
Data are given as the means±standard error of the mean (SEM). Student's t-test was used for comparison of pairs of differences, and statistics of P<0.05 were considered significant.
Results
Ezh2 expression increases during iPS cell generation
ES cells and iPS cells express various PcG components (Fig. 1A, B and Supplementary Fig. S1A, B). For example, ES cells and iPS cells derived from MEF, neural stem cells (NSC), and hematopoietic stem cells (HSC) express high levels of Ezh2. MEF and HSC, which were used for reprogramming, express low Ezh2 levels. NSC contain rather high levels of Ezh2, which might be related to their high proliferation potential. Additionally, NSC are also readily reprogrammed into iPS cells by two factors (Oct4 and Klf4) only [28]. iPS cell lines differed in Ezh2 expression, which was related to the cell type used for reprogramming and/or due to clonal variation. Western blot analysis extended the Ezh2 RNA expression data and showed very low Ezh2 expression in MEF and high Ezh2 expression in ES cells and iPS cells (Fig. 1B).
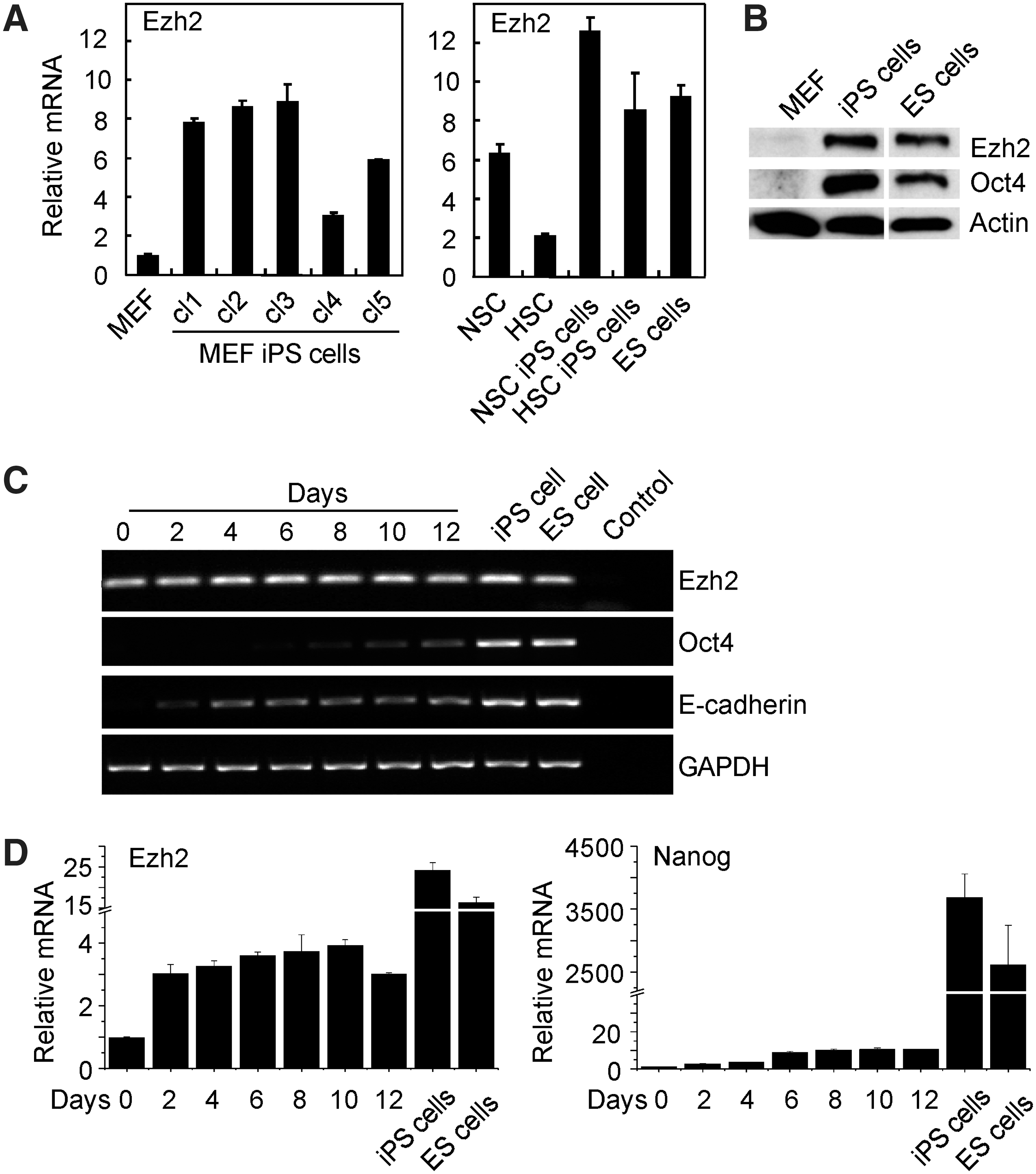
Ezh2 expression is induced during induced pluripotent stem (iPS) cell generation.
iPS cell generation entails the repression of somatic genes and activation of pluripotency genes, such as Oct4 and Nanog. To measure Ezh2 expression during reprogramming, MEF were transfected with the reprogramming factors, Oct4, Sox2, Klf4, and c-Myc (OSKM), and RNA was isolated in regular time intervals. The kinetics of Ezh2 expression was analyzed by RT-PCR and related to the expression of Oct4, Nanog, and E-cadherin (Fig. 1C, D). Ezh2 mRNA is already expressed in MEF and further increased during reprogramming, and thus, established iPS cell lines showed high Ezh2 expression. E-cadherin, an early marker for reprogramming, was already observed at day 2, whereas the pluirpotency markers Oct4 and Nanog were detected from day 6–8 onward.
These data indicate that during iPS cell generation Ezh2 expression is induced and its expression is high in established iPS cells. The increase of Ezh2 expression during reprogramming led us to investigate whether Ezh2 plays an active role in iPS cell generation.
Forced Ezh2 expression enhances reprogramming efficiency
To investigate the impact of forced Ezh2 expression on iPS cell generation, Oct4-eGFP MEF from OG2 mice [25] were infected with OSKM reprogramming factors plus Ezh2 using retroviral vectors. Infected cells were maintained in the ES cell culture medium, and Oct4-GFP+ colonies appeared around day 15 after infection, both with and without Ezh2 (Fig. 2A). Interestingly, forced Ezh2 expression caused about 1.5- to 2-fold higher frequency of Oct4-GFP+ cell and iPS cell colonies (Fig. 2A–D). The increase in iPS cell colony formation did, however, not reach statistical significance, probably due to variation in colony formation, which is frequently observed for primary iPS cells. Ezh2-iPS cells displayed typical ES cell morphology and growth characteristics and expressed the pluripotency markers, Oct4, Sox2, Nanog, Dppa4, and SSEA1 (Fig. 2E, F). Finally, Ezh2-iPS cells were injected into NOD-SCID mice and analyzed for teratoma formation. Ezh2-iPS cells developed teratomas in recipient mice, which were of comparable size as those of ES cells and iPS cells without Ezh2 (data not shown). Histologically, these teratomas contained cell derivatives of all three germ layers (Supplementary Fig. S2).
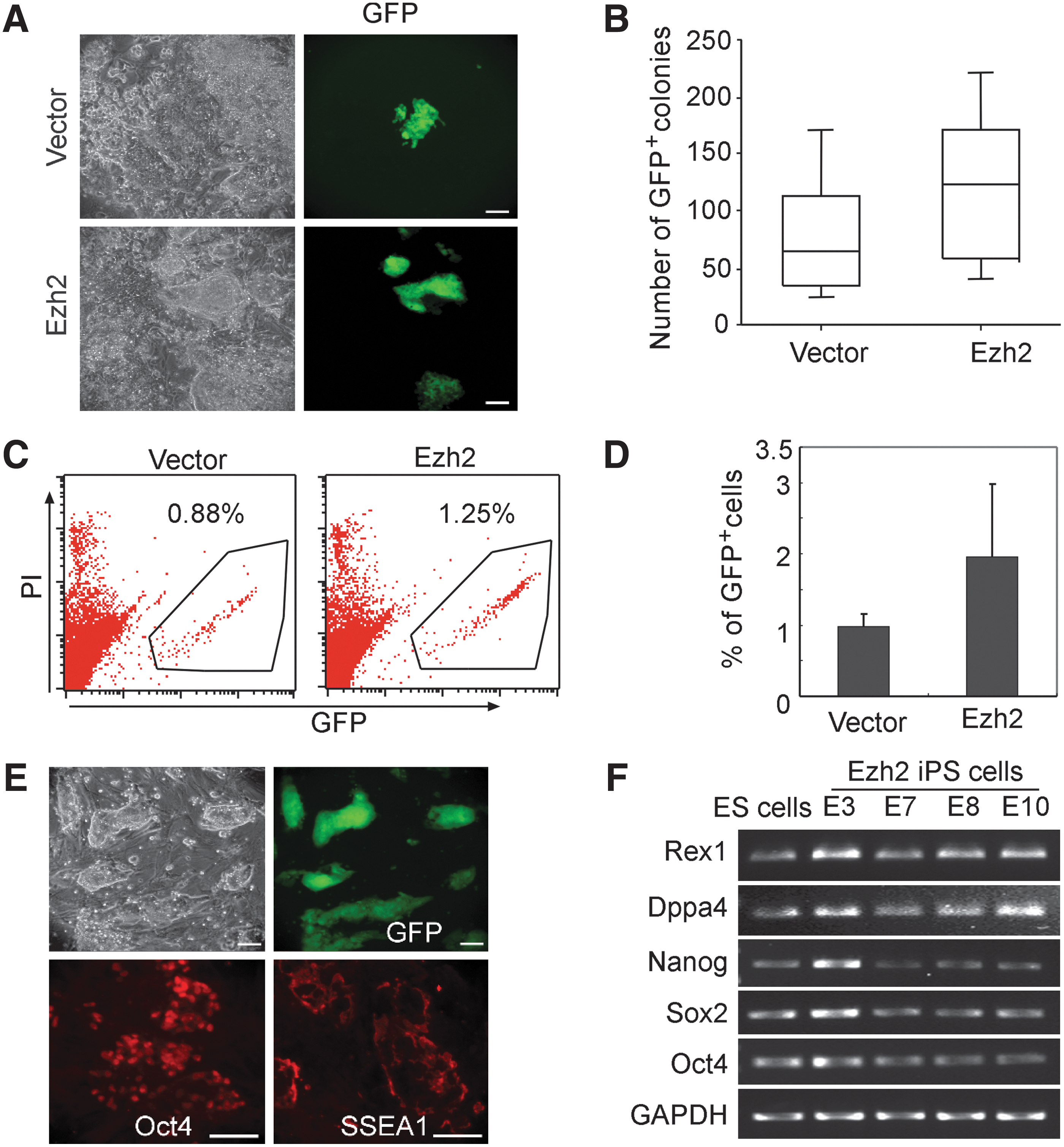
Forced Ezh2 expression enhances the efficiency of iPS cell generation. Generation of iPS cells from Oct4-eGPF OG2 MEF with forced Ezh2 expression. OG2 MEF were transduced with the four reprogramming factors (Oct4, Sox2, Klf4, and c-Myc, OSKM) plus Ezh2, or empty vector control (vector).
In summary, forced Ezh2 expression enhanced the four-factor induced iPS cell generation to some extent, and such Ezh2-iPS cells showed biological properties similar to iPS cells without ectopic Ezh2 expression.
c-Myc induces Ezh2 expression, but Ezh2 cannot compensate for c-Myc in iPS cell generation
MEF lose their proliferative capacity with extended periods of time in culture and acquire a senescent phenotype, which is accompanied with the loss of Ezh2 expression [35]. shRNA-mediated Ezh2 downregulation also induces senescence (Supplementary Fig. S3D). Conversely, ectopic expression of Ezh2 overcomes senescence in MEF [35]. iPS cells show high Ezh2 expression (Fig. 1A–D) and also overcome senescence [36]. Therefore, iPS cell generation appears to be linked to high Ezh2 expression. Thus, to determine which reprogramming factor induces Ezh2 during iPS cell generation, MEF cells were transduced with individual reprogramming factors (Oct4, Sox2, Klf4, or c-Myc), and Ezh2 mRNA was determined at day 6 by qRT-PCR. Ezh2 expression was significantly upregulated in c-Myc-transduced cells, whereas cells transduced with the four reprogramming factors showed highest Ezh2 expression (Fig. 3A).
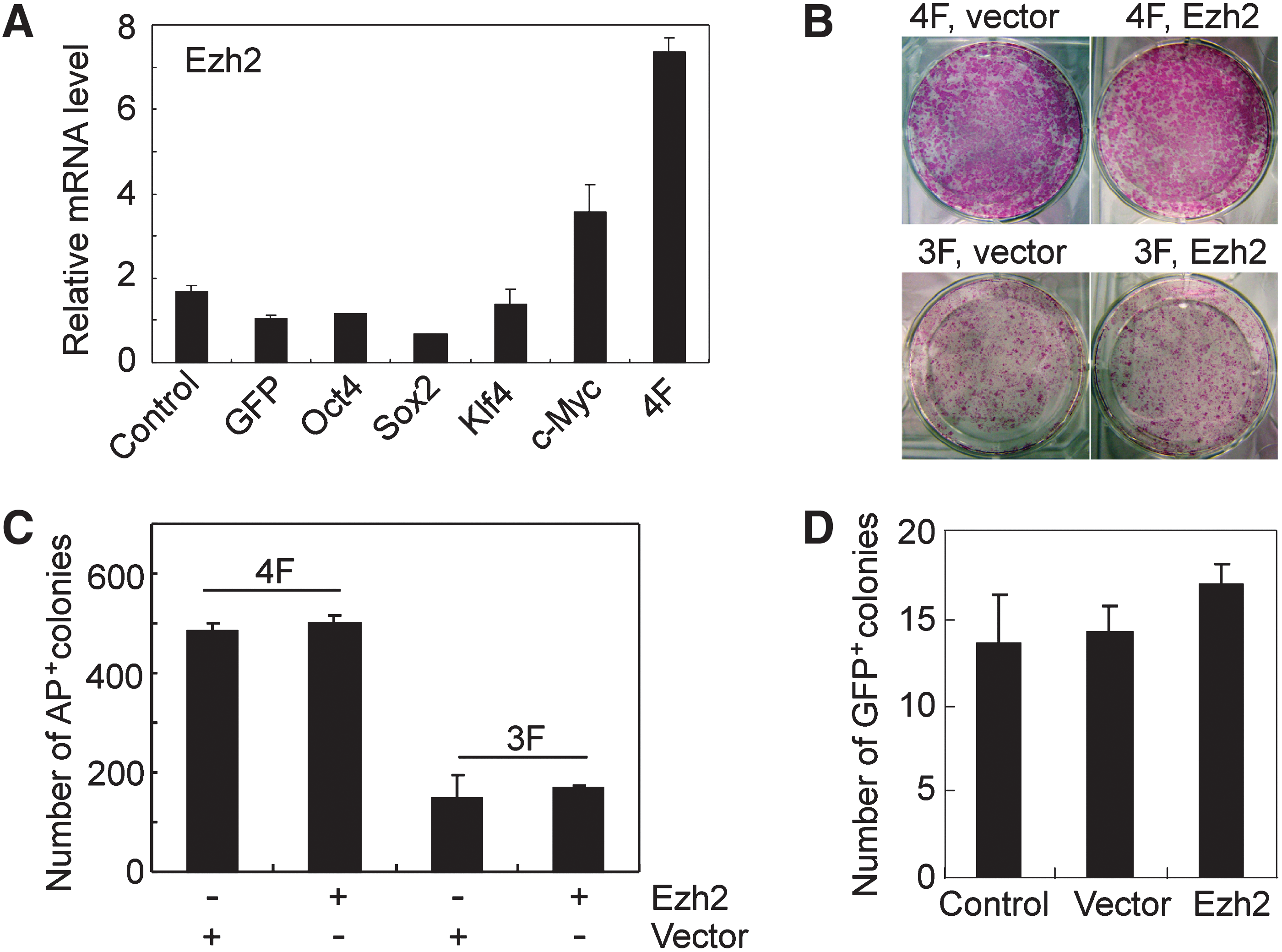
c-Myc induces Ezh2 expression, but Ezh2 cannot replace c-Myc during iPS cell generation.
c-Myc is important for efficient generation of iPS cells, and thus, the question was whether forced expression of Ezh2 can substitute for c-Myc in iPS cell generation. To this end, iPS cells were generated from MEF with the three reprogramming factors, Oct4, Sox, and Klf4 (3F), with and without Ezh2 (Fig. 3B, C). Frequencies of alkaline phosphatase-positive (AP+) cells at day 10 after transduction were reduced without c-Myc, yet forced Ezh2 expression could not substitute for c-Myc (Fig. 3B, C). Essentially, the same result was obtained when Oct4-eGFP MEF of OG2 mice were used for reprogramming with Oct4, Sox2, and Klf4 (3F) with and without Ezh2. The frequencies of Oct4-GFP+ cells at day 25 after transduction were only slightly increased with forced Ezh2 (Fig. 3D).
Thus, c-Myc induces Ezh2 expression, yet forced Ezh2 expression cannot substitute for c-Myc in efficient iPS cell generation.
Ezh2 is required for efficient iPS cell generation
Ezh2 is important for establishing pluripotency. For example, (i) Ezh2-deficient mice do not allow the generation of ES cell lines or allow their generation only at very low efficiency and (ii) Ezh2-deficient ES cell lines failed in reprogramming B cells toward pluripotency by cell fusion [7,9]. In addition, as reported here, Ezh2 expression is robustly upregulated during iPS cell generation (Fig. 1A–D). Thus, we used shRNA-mediated knockdown of Ezh2 to determine whether Ezh2 is required for iPS cell generation. Lentivirus-mediated shRNA targeting of Ezh2 caused effective downregulation of Ezh2 expression in MEF, as determined by qRT-PCR (Supplementary Fig. S3A). Concomitantly with Ezh2 knockdown, MEF ceased proliferating, showed a lower frequency of cells in S+G2 phase, and acquired a senescent phenotype (Supplementary Fig. S3B–D).
To determine the impact of Ezh2 on iPS cell generation, MEF were infected with retroviruses containing the four reprogramming factors OSKM together with either Ezh2 shRNA hairpins (shEzh2-1, shEzh2-2) or control shRNA (shLuc; Fig. 4A, B). Ezh2 expression was efficiently silenced by Ezh2 shRNA, which led to a reduction in global H3K27me3 levels as demonstrated by western blotting (data not shown). At day 10, cells were stained for AP, which represents an early marker for iPS cell generation. Cells infected with shLuc control generated frequencies of AP+ colonies similar to control. Importantly, Ezh2 downregulation caused a strong reduction of AP+ colonies (Fig. 4A, B). At day 20, we scored cultures for iPS cell colonies by morphology and observed a prominent reduction in iPS cell colony numbers by shEzh2-1 and shEzh2-2 compared to controls (Fig. 4C).
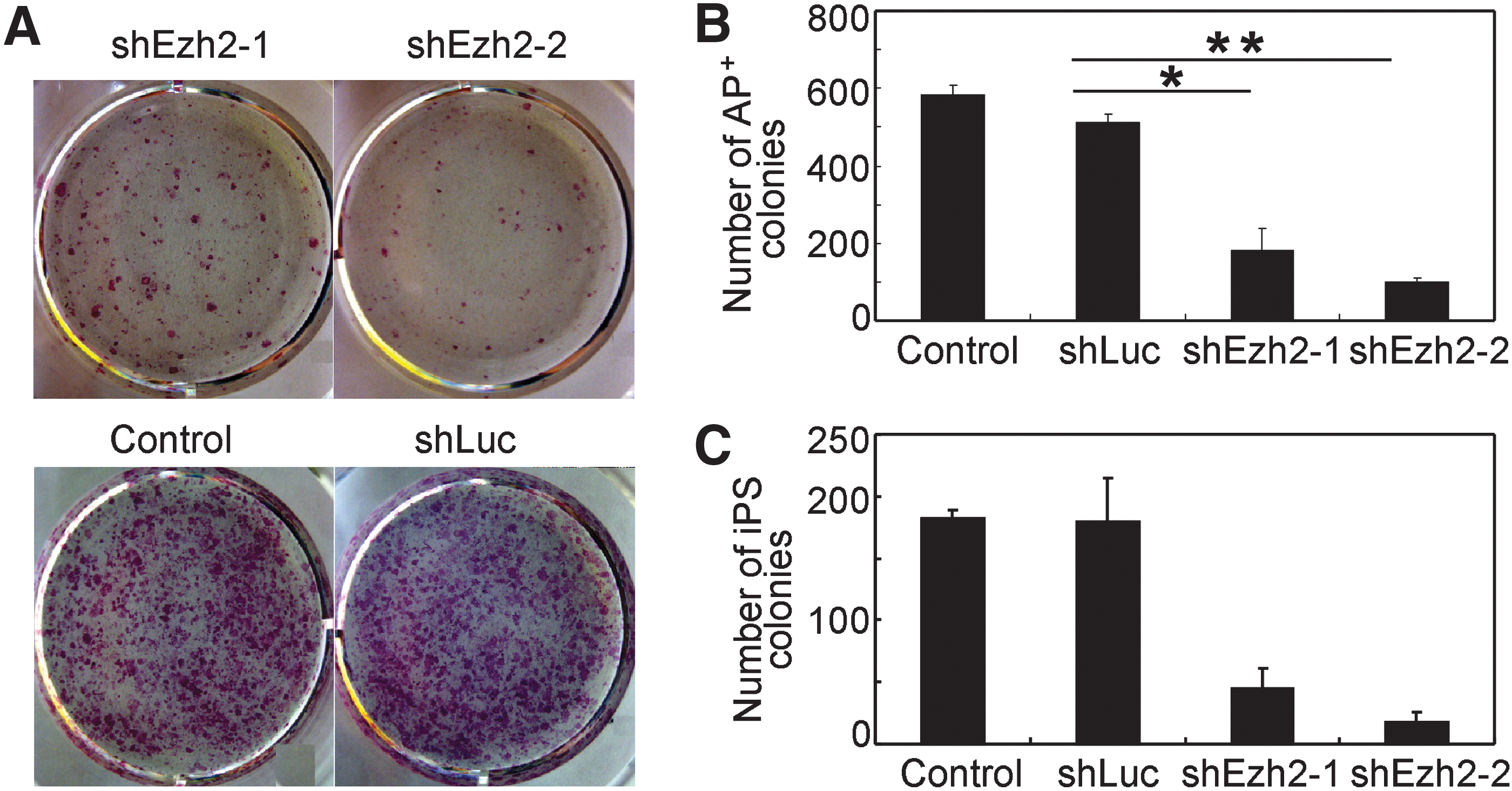
Knockdown of Ezh2 expression impairs iPS cell generation.
We next wanted to clarify why some iPS cells developed and whether these were refractory to shRNA-mediated Ezh2 knockdown. Therefore, iPS cells were generated with OSKM retroviruses together with either shEzh2 GFP vector (shEzh2-1, shEzh2-2) or control shLuc GFP vector, as described above. A panel of iPS cell clones refractory to shEzh2 was isolated and expanded. All iPS cell clones generated in the presence of shEzh2 GFP vector were GFP negative by fluorescence microscopy, indicating that they did not take up or express shEzh2 GFP vector and thus escaped Ezh2 knockdown. iPS cell clones with shLuc GFP vector were GFP+. Next DNA and RNA were isolated and analyzed for shEzh2 GFP vector sequences and vector expression by PCR. We found that all iPS cell clones that escaped shRNA-mediated Ezh2 knockdown did either not contain vector sequences or expressed vector sequences at very low levels (Supplementary Fig. S4A, B). Additionally, all iPS cell clones expressed Ezh2 mRNA at levels, which were similar to those in 4F iPS cells without shEzh2 or with shLuc control, or in ES cells. There was no shRNA-mediated knockdown of Ezh2 in all shRNA iPS cell clones analyzed, whereas we observed efficient shRNA-mediated knockdown of Ezh2 in MEF control (Supplementary Fig. S4C). Thus, there are apparently two reasons for escape of Ezh2 knockdown: (i) the lack of successful infection with shEzh2 vector and (ii) low and most probably insufficient expression of shEzh2 vector. Taken together, knockdown of Ezh2 severely impaired iPS cell generation, which shows that Ezh2 is required for efficient iPS cell generation.
We then proceeded to investigate whether Ezh2 also impacts on iPS cell self-renewal. shEzh2-1 and shEzh2-2 caused effective knockdown of Ezh2 in iPS cells and ES cells, and a reduction in H3K27me3 and an increase of the PRC2 target gene Gata4 (Supplementary Fig. S5A, B). Yet knockdown of Ezh2 did not affect (i) the characteristic colony morphology of iPS cells and ES cells, (ii) expression of the pluripotency markers Oct4 and SSEA1, and (iii) proliferation of iPS cells and ES cells (Supplementary Fig. S5B–D, and data not shown). Thus, Ezh2 is dispensable for ES cell and iPS cell self-renewal in culture.
Ezh2 acts in iPS cell generation through repressing the Ink4a/Arf locus
Ezh2 represses the Ink4a/Arf locus, which encodes the cell cycle kinase (CDK) inhibitors p16Ink4a and p19Arf [37]. In addition, Ezh2 overexpression allows MEF to bypass senescence by repressing the Ink4a/Arf locus (Supplementary Fig. S6A–C) [35,37]. During iPS cell generation, the Ink4a/Arf locus is epigenetically reprogrammed toward a bivalent silent configuration with H3K27me3 and H3K4me3 marks, which leads to Ink4a/Arf repression [30].
Ezh2 knockdown increases Ink4a expression in ES cells and iPS cells and during iPS cell generation (Supplementary Fig. S7A, B). Therefore, we investigated whether Ink4a/Arf knockdown can rescue the impaired iPS cell generation by Ezh2 knockdown and performed simultaneous Ezh2 and Ink4a/Arf knockdown experiments. MEF were cotransfected with reprogramming factors, OSKM and Ezh2 shRNA, and with or without shRNA targeting Ink4a/Arf. Efficiency of reprogramming was measured at day 10 by AP staining (Fig. 5A, B). In accord with published data [30], we observed a higher frequency of AP+ colonies with OSKM and shInk4a/Arf. As expected, Ezh2 knockdown increased Ink4a expression and impaired iPS cell generation (Supplementary Fig. S7B and Fig. 4A, B), and this was partially rescued by simultaneous knockdown of Ink4a/Arf expression (Fig. 5A, B).
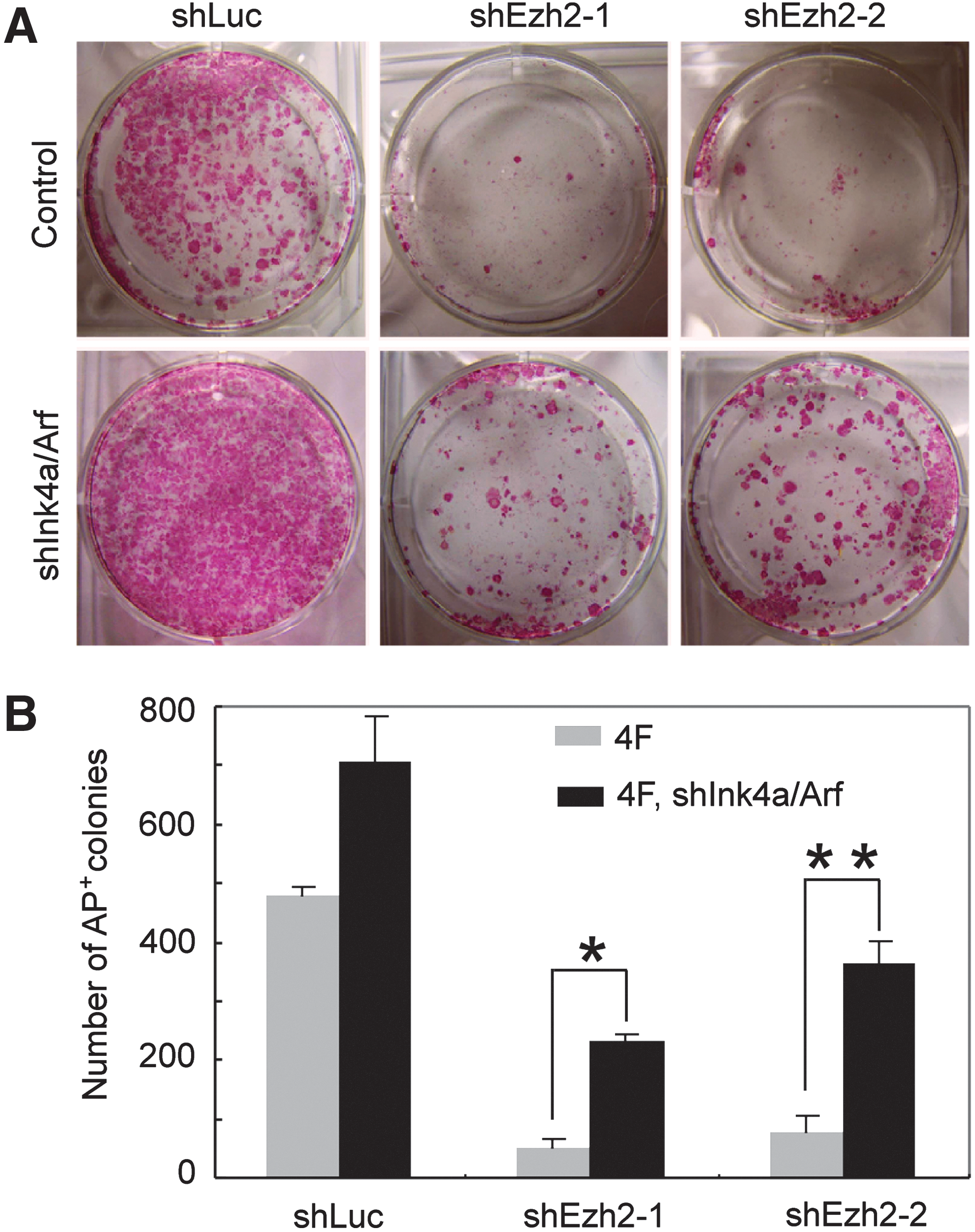
Ezh2 impacts on iPS cell generation in part through repressing Ink4a/Arf.
Discussion
In this study, we examined the impact of the PcG protein Ezh2 on iPS cell generation. We found that Ezh2 is induced during four-factor mediated reprogramming of MEF and that iPS cells contain high levels of Ezh2 protein, which are similar to the levels in ES cells. Importantly, Ezh2 knockdown severely impaired iPS cell generation, and thus, Ezh2 upregulation is critical for efficient reprogramming. We also found that Ezh2 impacts on iPS cell generation at least in part through repression of the CDK inhibitor Ink4a/Arf, which represents a major roadblock for iPS cell generation.
Somatic cell reprogramming and induction of pluripotency involve extensive chromatin reorganisation [15,16], and enforced expression of subunits of the chromatin remodeling BAF complex facilitates iPS cell generation [21]. Additionally, expression of Trithorax complex component WD repeat domain 5 (Wdr5), an effector of histone H3 lysine 4 (H3K4) methylation, is required for efficient iPS cell generation [38]. Furthermore, inhibition of the H3 lysine 79 (H3K79) methyltransferase DOT1L accelerates reprogramming and increases the frequency of iPS cells [22]. Thus, all these findings demonstrate the impact of epigenetic modifiers as cofactors of the OSKM reprogramming factors to repress somatic gene expression and to activate the pluripotency program [15,16,39].
Previous work showed that Ezh2 is important for establishing ES cell lines from blastocysts [7]. Additionally, Ezh2 was required for efficient somatic cell reprogramming by cell fusion and nuclear transfer [9,10]. These findings are in line with the results reported here. Ezh2 is abundantly expressed in iPS cells, and Ezh2 knockdown severely impaired iPS cell generation. However, once pluripotency is established, Ezh2 knockdown leaves the pluripotent phenotype of iPS cells unaffected. Additionally, as expected, Ezh2 knockdown reduced H3K27 trimethylation. But some H3K27me3 remained, probably due to compensation by other methyltransferases, such as Ezh1. Functional redundancy between Ezh1 and Ezh2 was described before [8,40]. All this indicates that Ezh2 is critical for induction of pluripotency, but once pluripotency is established, Ezh2 is not required anymore. The Ink4a/Arf locus represents a major target of Ezh2 [37] and repression of Ink4a/Arf improves iPS cell generation, and low Ink4a/Arf expression represent a feature of iPS cells and ES cells [30,41]. Thus, it appears conceivable that in established iPS cells Ink4a/Arf is silenced by mechanisms other than involving Ezh2. Yet, proper differentiation of iPS cells requires Ezh2 [42].
In a recent paper, Fragola et al. [24] generated iPS cells from MEF with a conditional Ezh2 knockout allele for the deletion of the catalytic Ezh2 SET domain. A cell-permeable TAT-Cre recombinase was used to inactivate Ezh2. Ezh2-deficient iPS cells were isolated, which exhibited a global loss of H3K27me3, yet a typical iPS cell phenotype, including ES cell-like morphology, growth, and differentiation potential. These findings contrast previous reports on Ezh2-deficient blastocyts to produce no or very few ES cells and on Ezh2-deficient ES cells to fail in cell fusion-induced reprogramming of somatic cells toward pluripotency [7 –9]. The results by Fragola et al. are also in contrast to the shRNA knockdown studies reported here on primary iPS cells and on a doxycylin-inducible secondary iPS cell system [17]. Additionally, it also contrasts findings by the same authors on shRNA-induced knockdown of the PRC2 component EED, which reduced iPS cell generation by over 80% [24]. Thus, the result on Ezh2-deficient iPS cells might be rather methodically, in that Ezh2 inactivation could have occurred after reprogramming. Such an interpretation would be in line with the results reported here on Ezh2 knockdown in established pluripotent cells, which leaves morphology, pluripotency, and self-renewal of ES cells and iPS cells unaffected.
PcG components show cell type-specific expression pattern [1 –3] and, as shown here, Ezh2 is abundantly expressed in ES cells and iPS cells. Elevated levels of Ezh2 expression have also been reported in tumor cells [43,44]. Additionally, c-Myc, one of the iPS cell inducing factors, was recently shown to directly regulate the Ezh2 expression and to be required for maintaining high Ezh2 expression in ES cells [45]. So far, among the four reprogramming factors, only c-Myc can efficiently induce Ezh2 expression. Yet, as shown here, enforced Ezh2 expression enhanced reprogramming efficiency only to some extent and could not compensate for the function of c-Myc in iPS cell generation.
The PRC1 component Bmi1 is an essential regulator for adult stem cells rather than ES cells [46]. In our previous work, we showed that forced Bmi1 expression promotes hematopoietic cell development from ES cells [26]. We also found that Bmi1 can efficiently substitute for c-Myc in iPS cell generation (unpublished data). In line with this, a recent study demonstrated that forced Bmi1 expression caused transdifferentiation of mouse fibroblasts into NSC-like cells and enhanced reprogramming efficiency and iPS cell generation [47]. However, enhanced iPS cell generation by enforced Bmi1 expression might be due to additional functions of Bmi1, besides of being a component of PRC1 [48]. All this indicates a differential impact of chromatin modifying enzymes, including Ezh2 and Bmi1, on iPS cell generation. In addition, the H3K39 demethylase Jhdm1a/1b represent yet another chromatin modifier that impacts on iPS cell generation [49].
Ezh2 is known as a senescence-preventing gene [37]. MEF undergo senescence in vitro, concomitantly with Ezh2 downregulation, loss of H3K27me3 of the Ink4a/Arf locus, Ink4a/Arf activation, and cell cycle arrest [37]. Here, we found that during iPS cell generation, Ezh2 expression is induced and iPS cells contain high levels of Ezh2 protein. Additionally, Ink4a/Arf, a major target of Ezh2 in adult cells, has been identified as a roadblock of iPS cell generation [30,41]. Thus, we propose that during reprogramming and induction of pluripotency, Ezh2 upregulation and thus H3K27me3 efficiently represses Ink4a/Arf, which is crucial for efficient iPS cell generation. This notion is further supported by our observation that Ezh2 knockdown increases Ink4a/Arf expression and impairs iPS cell generation. Notably, Ink4a/Arf knockdown at least partially resuscitates the impaired iPS cell generation by Ezh2 downregulation. Our results therefore suggest that a major activity of the Ezh2/Ink4a pathway on iPS cell generation might be to counteract senescence and thereby to affect the number of cells that are susceptible to reprogramming. The findings reported here extend previous knowledge of epigenetic modifiers impacting on iPS cell generation and highlight the intrinsic connection of reprogramming factors, epigenetic modifiers, and cell cycle regulators during the reprogramming process.
There is a significant variability of individual iPS cell clones in their differentiation potential. This is related to the (i) target cells, (ii) reprogramming factors and vector systems used for reprogramming, and (iii) further so far largely unknown factors [50]. In ES cells, many genes are marked with H3K27me3, which is critical for their differentiation potential [10]. In addition, ES cells with disrupted PcG protein expression and consequently altered H3K27me3 show impaired differentiation potential [5,51]. Similarly, a recent study showed that Ezh2-null iPS cells exhibit severe defects in differentiation [42]. Thus, Ezh2 level might represent an important marker for evaluating iPS cell quality.
In summary, the data presented here demonstrate that Ezh2 impacts on iPS cell generation and acts at least in part through repressing the Ink4a/Arf locus, a major roadblock for iPS cell generation. The study emphasizes the importance of chromatin remodeling in cellular reprogramming toward pluripotency. In addition, it adds to our understanding of the underlying molecular mechanisms ongoing during iPS cell induction.
Footnotes
Acknowledgments
We thank Drs. A. Iwama and M. Serrano for plasmids. We also thank Andrea Offergeld and Renate Sous for expert secretarial assistance. This work was supported in part by grants from the Interdisciplinary Center for Clinical Research IZKF Aachen within the Faculty of Medicine, RWTH Aachen University, and DFG SPP1356 to M.Z. X.W. is a recipient of a fellowship of China Scholarship Council (CSC).
Author Disclosure Statement
No competing financial interestes exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.