
Abstract
With an incidence of ∼1:3,500 to 5,000 in male children, Duchenne muscular dystrophy (DMD) is an X-linked disorder in which progressive muscle degeneration occurs and affected boys usually die in their twenties or thirties. Cardiac involvement occurs in 90% of patients and heart failure accounts for up to 40% of deaths. To enable new therapeutics such as gene therapy and exon skipping to be tested in human cardiomyocytes, we produced human induced pluripotent stem cells (hiPSC) from seven patients harboring mutations across the DMD gene. Mutations were retained during differentiation and analysis indicated the cardiomyocytes showed a dystrophic gene expression profile. Antisense oligonucleotide-mediated skipping of exon 51 restored dystrophin expression to ∼30% of normal levels in hiPSC-cardiomyocytes carrying exon 47–50 or 48–50 deletions. Alternatively, delivery of a dystrophin minigene to cardiomyocytes with a deletion in exon 35 or a point mutation in exon 70 allowed expression levels similar to those seen in healthy cells. This demonstrates that DMD hiPSC-cardiomyocytes provide a novel tool to evaluate whether new therapeutics can restore dystrophin expression in the heart.
Introduction
D
In some instances, skipping does not produce a functional truncated protein. This is true when mutations lie in nonredundant regions of dystrophin, such as the N-terminal actin-binding domain or the C-terminal β-dystroglycan/syntrophin binding region. In such cases, early phase I/II clinical trials have been initiated to test efficacy of delivery of dystrophin minigenes to dystrophic muscle.
The focus of clinical trials has been primarily on evaluating the effect of treatments on skeletal muscle [7,8]. However, with over a third of DMD patients dying of heart failure, there is an urgent need for methods that evaluate whether therapeutics can rescue dystrophin expression in cardiomyocytes. Notably, the impact of skeletal muscle treatment on cardiac disease in DMD patients is under debate, with some authors suggesting benefit to the heart and others detriment [10]. In the latter case, it may be anticipated that improvements in assisted ventilation will correspond to an increasing number of patients dying due to heart failure in the future.
The skeletal muscle clinical trials have benefitted from numerous background studies in which therapeutics have been tested in cultured muscle cells derived from DMD patients [11]. Such in vitro work has allowed the efficacy of molecular treatments on human mutations and the patient's own cells to be determined [12]. However, there is no parallel in vitro system for human cardiomyocytes because primary human cardiomyocytes do not divide in culture and experiments cannot be carried out repeatedly on identical cultures. Moreover, primary human cardiomyocytes are obtained through invasive procedures (e.g., heart biopsies), thereby potentially putting patients at risk. An in vitro system that parallels that available for skeletal muscle would be beneficial, especially since there are considerable differences between skeletal and cardiac muscle, including physiology, drug-induced toxicity, and dystrophin expression and localization. Moreover, the two tissues differ in RNA production rate, mRNA levels and mRNA turnover. Half-life of skipping drugs is longer in the heart but delivery to cardiomyocytes is less efficient that skeletal muscle cells. This is because the membrane of DMD skeletal muscle is more leaky than cardiomyocytes, thereby allowing better uptake of skipping drugs [13]. Dystrophin levels are higher in the heart relative to skeletal muscle (Aartsma-Rus, submitted). These issues could alter the ability to restore dystrophin expression in the heart. Therefore, access to human cardiomyocytes with various DMD mutations would accelerate preclinical development of therapeutics.
In this article, we have used epigenetic reprogramming to produce human induced pluripotent stem cells (hiPSCs) from skin fibroblasts derived from seven DMD patients who harbor frame-shifting deletions or nonsense point mutations in different regions of the DMD gene (5′ end, middle or 3′ end). The pluripotent cells were induced to differentiate into electrophysiologically- and pharmacologically-functional cardiomyocytes that displayed a dystrophic gene expression profile. Delivery of AONs directed to exon 51 of the DMD gene induced exon skipping in DMD hiPSC-cardiomyocytes, leading to expression of truncated RNA transcripts that produced dystrophin protein to a level of ∼30% of wild-type cells. Viral transduction of DMD hiPSC-cardiomyocytes led to expression of a dystrophin minigene at the RNA and protein level to up to 90% of normal dystrophin levels. Therefore, the hiPSC-cardiomyocyte model can be used to evaluate the efficacy of novel therapeutics for DMD.
Materials and Methods
Production, maintenance, and differentiation of hPSC lines
Following informed consent under regulation of local ethics committee approval 08/H0906/28, human fibroblasts were prepared from 4 mm skin punch biopsies [14]. Fibroblast outgrowths were stored in the BioBank at the MRC centre for Neuromuscular Diseases in Newcastle. Induction of pluripotency, and establishment and characterization of the hiPSC lines was using the methods within references [15,16]. Culture of hESC (HUES7) and hiPSC lines (DMD 4, 7, 11, 15, 16, 19, 21, and a control hiPSC line, HF-hiPSC [14]) was on Matrigel, in mouse embryonic fibroblast-conditioned medium [17,18]. Experiments were carried out on hiPSC that were between passage 15 and 30. Differentiation into cardiomyocytes was by forced aggregation cells to form embryoid bodies (EBs) [17,18].
Culture of human primary myoblasts and differentiation into myotubes
Human myoblast cultures were derived from muscle biopsy samples and were obtained from the BioBank. These cells were cultured in skeletal muscle cell growth medium, SMGM (Promocell), supplemented with 10% foetal calf serum (Gibco), 4.5 mM GlutaMAX™ (Invitrogen), and 50 μg/mL Gentamicin (Gibco). Cells were maintained at subconfluence over 2–5 passages. For differentiation, myoblasts were seeded into six well plates according to their growth rate and allowed to reach 70% confluence over 2–3 days. At this point, medium was changed to myoblast differentiation medium, containing high glucose DMEM (Gibco) supplemented with 5% heat-inactivated horse serum (Sigma-Aldrich). After 4–10 days the morphology of the cells changes and cells fuse to form myotubes.
Immunocytochemistry analysis of DMD hiPSC-cardiomyocytes
Beating clusters of cardiomyocytes were dissociated into single cells using a three step method [17]. For cardiac α-actinin staining, cells were washed with phosphate-buffered saline (PBS) then fixed in 4% w/v paraformaldehyde and stained [14]. For detection of dystrophin, cells were washed with PBS and fixed in ice-cold 100% methanol for 1 min. Cells were then washed with PBS, permeabilized and stained following the same method as above. The antibodies used for detection of dystrophin were Dys-C (C-terminus, Novocastra 1:10), Dys-Rod (rod domain, Novocastra 1:10), Dys-N (N-terminus, Novocastra 1:10), and MANDYS1 (recognizes epitope in exon 31–32 of dystrophin; kind gift from Glenn Morris, 1:10). The antibody for δ-sarcoglycan was purchased from Novacastra and used at a dilution of 1:10. Detection was with a goat anti-mouse Cy3 secondary antibody (Vector Labs 1:500). Cells were counterstained with a rabbit anti-cardiac troponin I antibody (Abcam 1:200) and detected with a goat anti-rabbit AlexaFluor 488-conjgated secondary antibody (Molecular Probes 1:1000). Protein expression by immunofluorescence was quantified using Volocity software (version 5.2.0.0; PerkinElmer) to determine mean fluorescence intensity of each cell. For flow cytometry, dissociated cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton-X100, and then incubated with anti-cardiac troponin I (1:250) for 1 h at room temperature. The secondary antibody was Alexfluor-488 (1:250; Vector Labs), with staining for 1 h at room temperature. Cells were then washed in PBS two times before analyzing using the Beckman Coulter FC500 flow cytometer with FlowJo software (Treestar).
TaqMan quantitative real-time PCR
TaqMan quantitative PCR was carried out using Applied Biosystems Assay on Demand primers/probe sets to different isoforms of the DMD gene. DMD hiPSCs were differentiated as EBs for 30 days and RNA was collected for analysis. Differentiated DMD hiPSCs were compared to the differentiated control hiPSCs. Expression of the assays detecting all isoforms (Hs00758087_m1), exons 47–48 (Hs01049444_m1), or Dp427m specific (Hs01049418_m1) mRNA, was quantified using GAPDH as an internal control (Hs99999905_m1). Assays were carried out using Applied Biosystems 7500 Fast Real-time PCR System and software.
Electrophysiology analysis by patch clamp and microelectrode array
For electrophysiological extracellular field potential measurements [14,17], beating clusters were directly seeded onto Matrigel-coated microelectrode arrays (Scientifica). For pharmacological analysis, a 5 min basal reading was recorded, followed by 5 min intervals with increasing doses of isoprenaline, from 1–1000 nM. After the final dose, beating clusters were washed in medium and allowed to recover for 20 min, before taking a final “washout” reading. For single cell electrophysiology, whole-cell patch clamp was carried out on dissociated cells, analyzed in normal current-clamp mode [14]. All electrophysiology data were analyzed in Axon™ pClamp® 9.0 software.
Exon skipping in DMD hiPSC-cardiomyocytes and myotubes
For exon-skipping experiments, myotubes, beating clusters or dissociated cardiomyocytes were seeded onto 35 mm MatTek dishes with glass coverslip inserts (MatTek) and allowed to recover for 3–6 days. Cells were transfected with polyethylenimine (PEI) (ExGen500; MBI Fermentas) in Opti-MEM reduced serum medium (Invitrogen) for 3 h, according to the manufacturer's instructions. For these experiments 3.5 μL PEI was used per μg anti-sense oligonucleotide (AON) [11]. For disaggregated cardiomyocytes, a concentration of 100 nM was used, whereas for beating clusters and myotubes 250 nM was used. Cells were transfected with a nontargeting or AON targeted to skip exon 51(h51AON). Exon skipping was analyzed by PCR using primers flanking the skipped area, from exon 43 to exon 54 (forward: 5′-gctcaggtcg gattgacatt-3′; reverse: 5′-tccttagctt ccagccattg-3′) or by protein analysis of MANDYS1 expression in hPSC-cardiomyocytes by immunofluorescence (cardiomyocytes were identified by reactivity with anti-cardiac troponin I antibodies).
Lentiviral production and transduction
A 4.5 kb dystrophin minigene was cloned into a second generation pSIN-IRES-blastocidin lentiviral vector. Virus was produced in the BL15 packaging cells, which have been engineered from HES293T line and allow purification of virus using conjugation with streptavidin paramagnetic beads. A full protocol can be found in reference [19].
Statistics
Statistics were performed using Prism Software (version 5.0 GraphPad).
Results
DMD patient selection
To address the need for an in vitro test-bed in human cells, we sought to produce hiPSC-cardiomyocytes from patients with different mutations in the DMD gene. We first performed isoform-specific reverse transcriptase–PCRs to identify the expression profile of dystrophin during differentiation of hPSCs to cardiomyocytes. Of the 18 potential isoforms produced via alternative splicing of transcripts produced from seven different promoters (Genbank Gene ID 1756; ncbi.nlm.nih.gov/gene/1756), only the short Dp71b isoform was expressed in undifferentiated cells (Fig. 1A). Differentiation to cardiomyocytes activated additional expression of the Dp427m muscle-specific long isoform and the intermediate Dp140 isoform (Fig. 1A). Therefore, we identified seven patients who had mutations (i) in the 5′ end of the gene, which disrupts only the Dp427m isoform (region 1; patients DMD11 and DMD 19), (ii) in the middle of the gene, which disrupts Dp427m and Dp140 (region 2; DMD4, DMD7, DMD 11, and DMD15), or (iii) in the 3′ end disrupting all three isoforms (region 3; DMD16) (Fig. 1B, C). Genotyping confirmed the presence of these mutations. Data were corroborated by histological and molecular analysis of muscle biopsies, and by clinical examination (Fig. 1D).
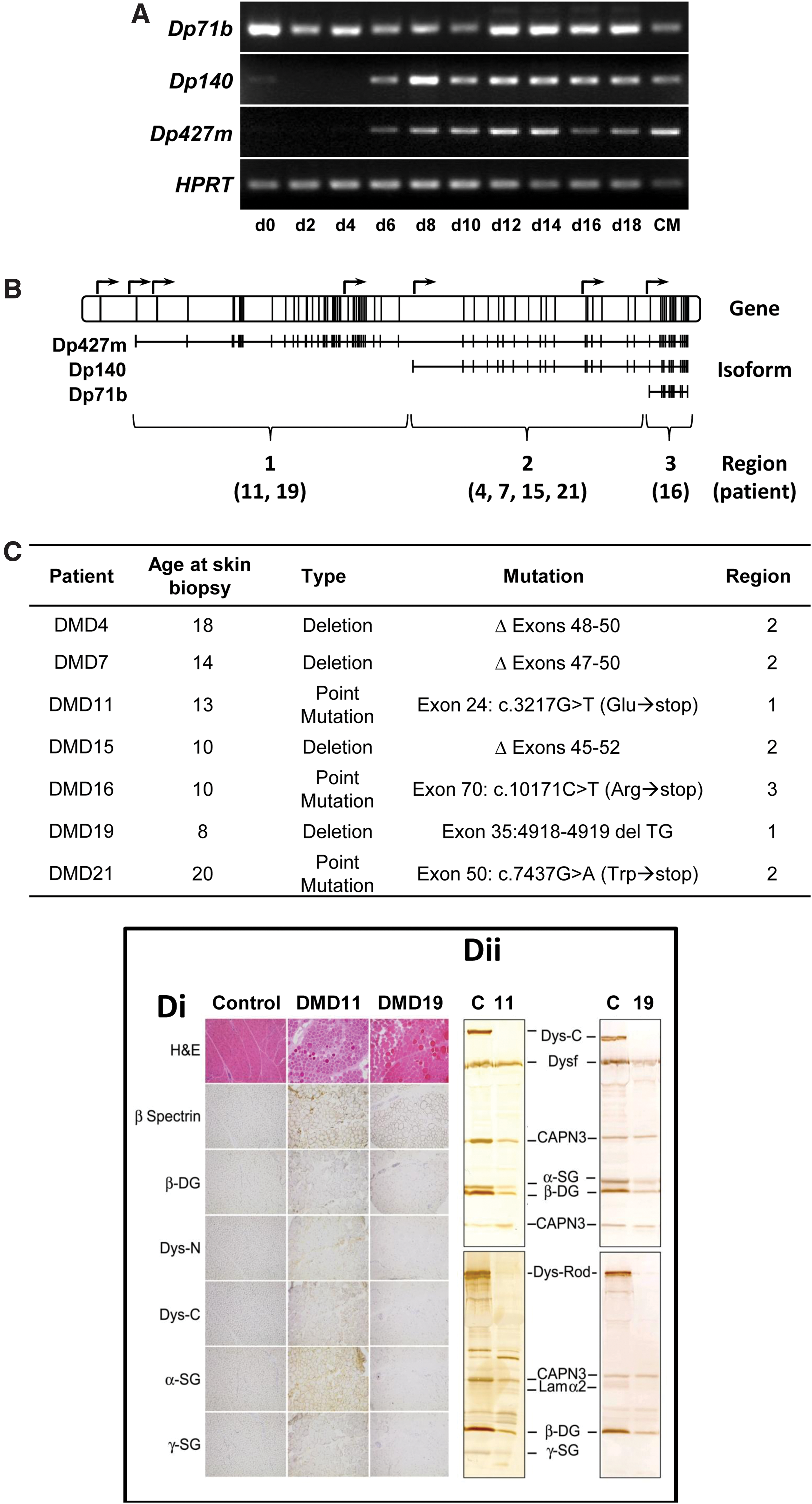
Selection of patients harboring DMD mutations. In
hiPSC production from DMD skin biopsies
Processing and culture of patient skin biopsies produced outgrowths that were transduced with four reprogramming factors [19,20], OCT4, SOX2, NANOG, and LIN28, which led to production of hiPSC over the next 3–4 weeks. hiPSC lines were established and characterization showed they conformed to accepted criteria [21] (Supplementary Fig. S1; Supplementary Data are available online at
DMD hiPSC-cardiomyocytes are functional
Production of spontaneously beating cardiomyocytes occurred from day 10 of differentiation of hiPSCs and there was no apparent difference in efficiency between the lines (data not shown). For all analyses, areas of beating cells were mechanically isolated to provide cardiomyocytes that could be reseeded as intact clusters or as single dissociated cells. This simple enrichment strategy produced cultures for analysis in which cardiomyocyte purity was approximately 85%, as determined by flow cytometry of cells stained with cardiac troponin I (Fig. 2A). The remaining cells were fibroblasts on account of their reactivity with the fibroblast-specific marker, P4HB (prolyl 4-hydroxylase, beta polypeptide; data not shown). hPSC-cardiomyocytes were charactersized by immunostaining with α-actinin, which showed sarcomeric striations typical of these cells (Fig. 2B). Whole cell patch clamp electrophysiology indicated presence of atrial-, ventricular-, and pacemaker-like cardiac subtypes (Fig. 2C), while extracellular field potentials could be detected by multielectrode array analysis (Fig. 2D). Stimulation with escalating doses (1–1000 nM) of the β-adrenoceptor agonist, isoprenaline, led to increased beat rates (to 135%±14% at 1 μM) similar to those seen for HUES7 cells (P=0.38; t-test) and the chronotropic effect was reversed by drug washout. Therefore, the DMD-hiPSC lines could be differentiated into functional cardiomyocytes.
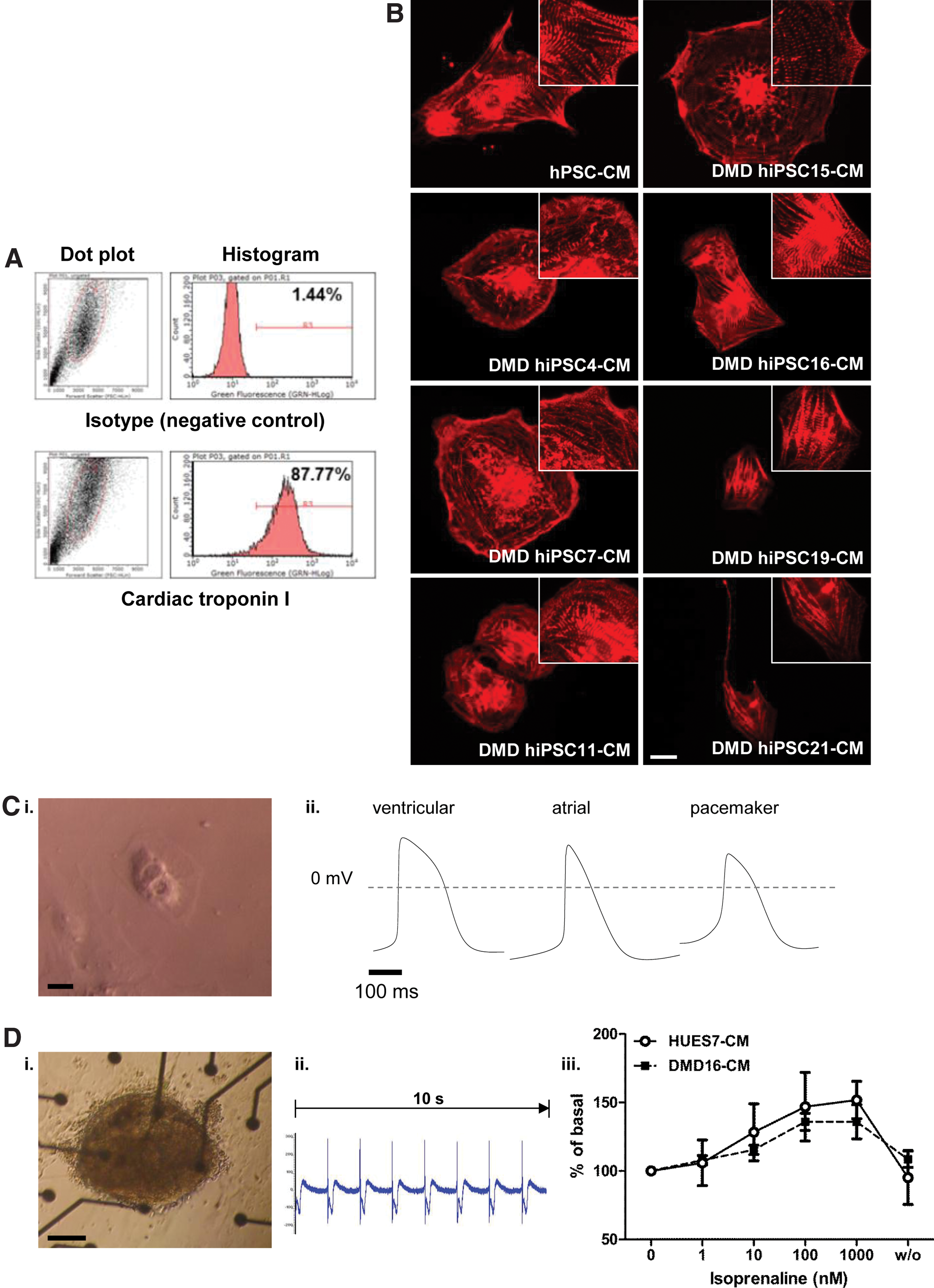
Cardiac differentiation of Duchenne muscular dystrophy (DMD) human induced pluripotent stem cells (hiPSC) lines.
Mutation profile is retained during differentiation of DMD hiPSCs
Since differentiation of the lines proved to be similar, we elected to use lines DMD hiPSC4 (deletion exons 48–50), DMD hiPSC7 (deletion exons 47–50), DMD hiPSC16 (point mutation in exon 70: c.3217G>T causing p.Trp1029×), and DMD19 (deletion of TG from exon 35) in subsequent studies. Lines 4 and 7 would be suitable for testing exon 51 skipping, while lines 16 and 19 would allow testing overexpression of a mini dystrophin construct.
To confirm the dystrophic gene expression status of these hiPSC lines, each was differentiated and examined by quantitative real-time PCR (Fig. 3). Assay 1, with primers located in regions 1 (exons 1–2, detects Dp427 muscle-specific isoform), showed significant reduction in expression in each of the hiPSC lines (Fig. 3B; P=< 0.01 to <0.001). Assay 2 (Region 2; exons 47–48, Dp427m and Dp140) detected significant reductions in gene expression for hiPSC lines, DMD4, DMD7, and DMD16 (P=<0.01 to <0.001). However, with the region 3 assay (exons 75–76, detects Dp427m, Dp140, and Dp71b), only DMD hiPSC16 cells had significantly reduced transcript levels (P=0.022), consistent with the exon 70 point mutation affecting all three isoforms (Dp427m, Dp140, and Dp71b).
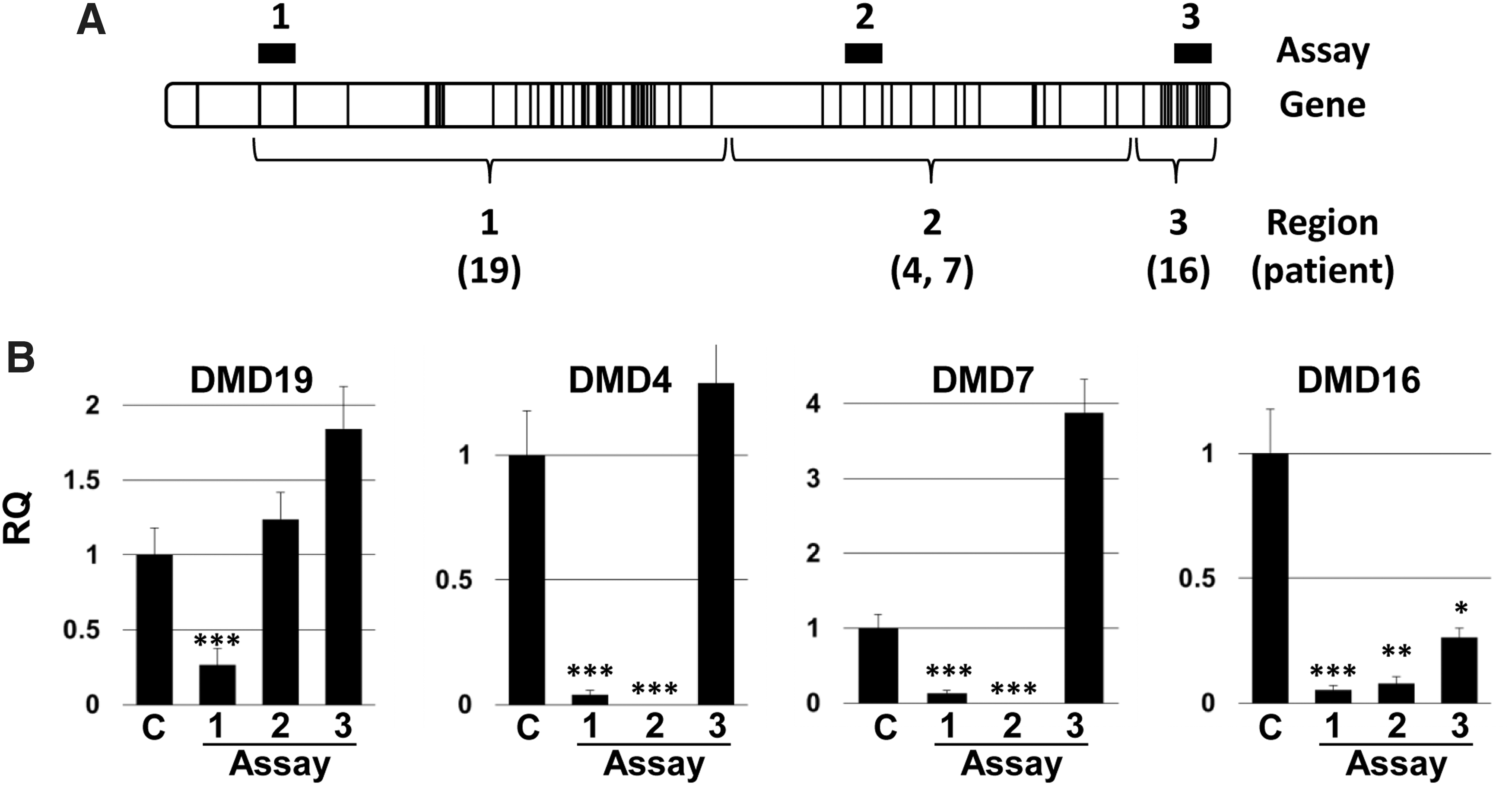
Gene expression analysis of DMD hiPSCs.
These data were also corroborated at the protein level, where an antibody to region 3 (C-terminus) detected expression in cardiomyocytes from DMD hiPSC4, DMD hiPSC7, and DMD hiPSC19 but not DMD hiPSC16 (Fig. 4). Dystrophin expression could not be detected in DMD hiPSC4, DMD hiPSC7, and DMD hiPSC16 using antibodies to region 2 (rod domain), while expression was not detected in any of the lines when probing region 1 (N-terminus). In all cases, staining with these antibodies showed dystrophin expression in normal myotubes or hPSC-cardiomyocytes in which DMD mutations were absent (Fig. 4). These data indicated that the gene expression profile of dystrophin in cells differentiated from the hiPSC lines matched the mutation status.
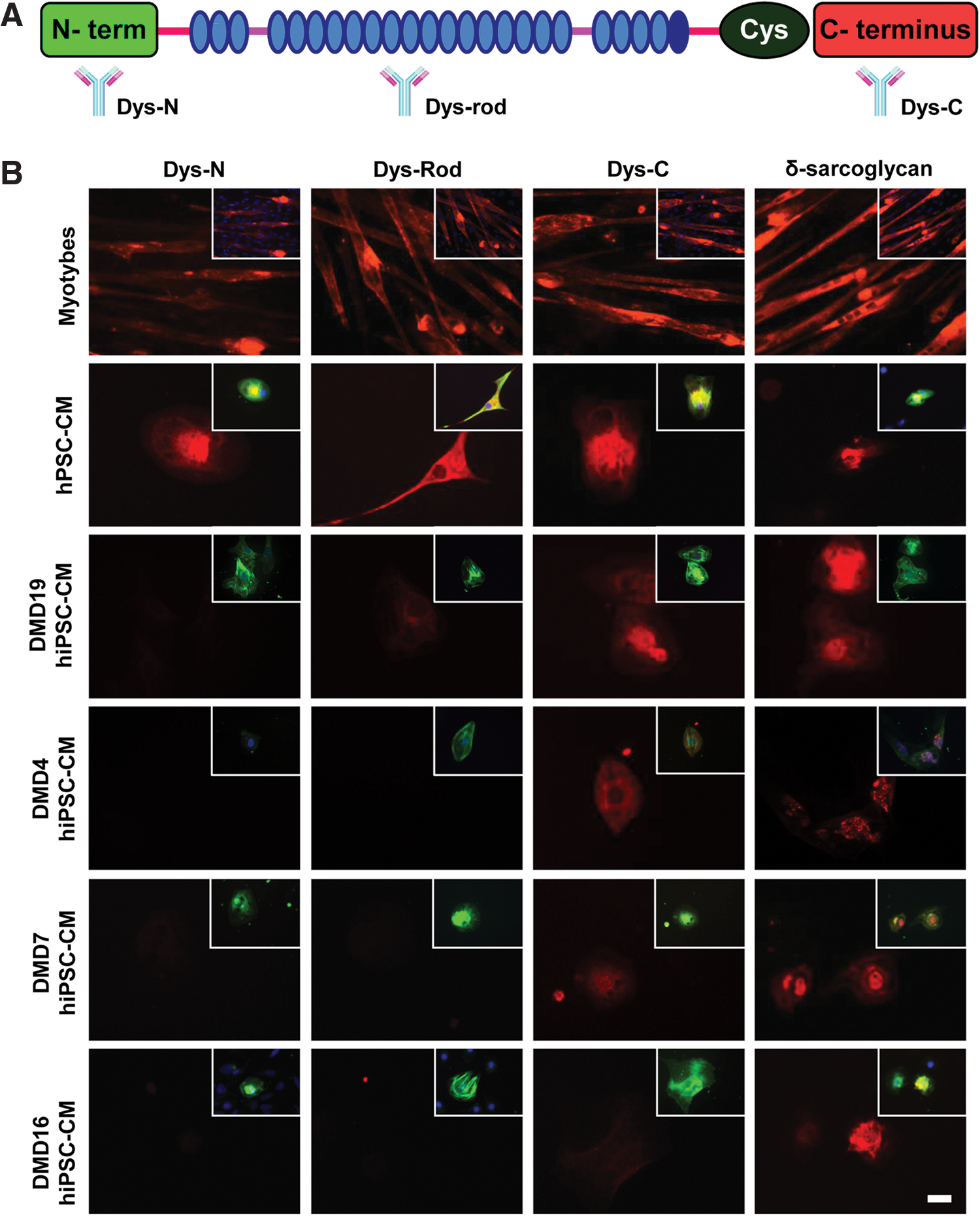
Protein expression in DMD hiPSCs. A schematic shows location of antibodies that detect N-terminus (Dys-N), rod domain (Dys-Rod), and C-terminus (Dys-C) of dystrophin
Genetic therapies facilitate dystrophin expression in DMD hiPSC-cardiomyocytes
Over 70% of DMD cases are caused by deletions of one or several exons of the dystrophin gene usually resulting in loss of the reading frame, and absence of dystrophin protein [23]. In particular, the central rod domain flanking exon 50 is a hot spot for deletions. This is typified by DMD hiPSC4 (exons 48–50) and DMD hiPSC7 (exons 47–50), which were selected as representative examples for evaluating exon-skipping drugs, particularly those for exon 51. Clinical trials are ongoing with both 2OMePS and PMOs (2′-O-methyl phosphorothioate and phosphorodiamidate morpholino oligos). Here, we employed 2OMePS, which are developed furthest. These drugs have been shown to function in patients and can be transfected at higher efficiencies than PMOs into myoblasts in vitro to induce exon skipping (Supplementary Fig. S2 and ref [24]).
Our initial attempts to transfect hiPSC-cardiomyocytes with fluorescently-tagged AONs were unsuccessful due to high toxicity and loss of cell viability. However, by reducing the concentration of the cationic lipid transfection reagent and optimizing timing and lipid:AON ratios, delivery to 90% of the hiPSC-cardiomyocytes was achieved (Supplementary Fig. S2). Transfection of DMD hiPSC-cardiomyocytes with exon 51 AONs caused exon skipping, and sequencing showed in-frame transcripts of exon 47–52 and 46–52 in lines 4 and 7, respectively (Fig. 5A, B). By day 3 after AON transfection, immunostaining showed dystrophin protein was approaching 30% of control cardiomyocytes (Fig. 5A–C), which corresponds to the level of rescue required for therapeutic benefit [25].
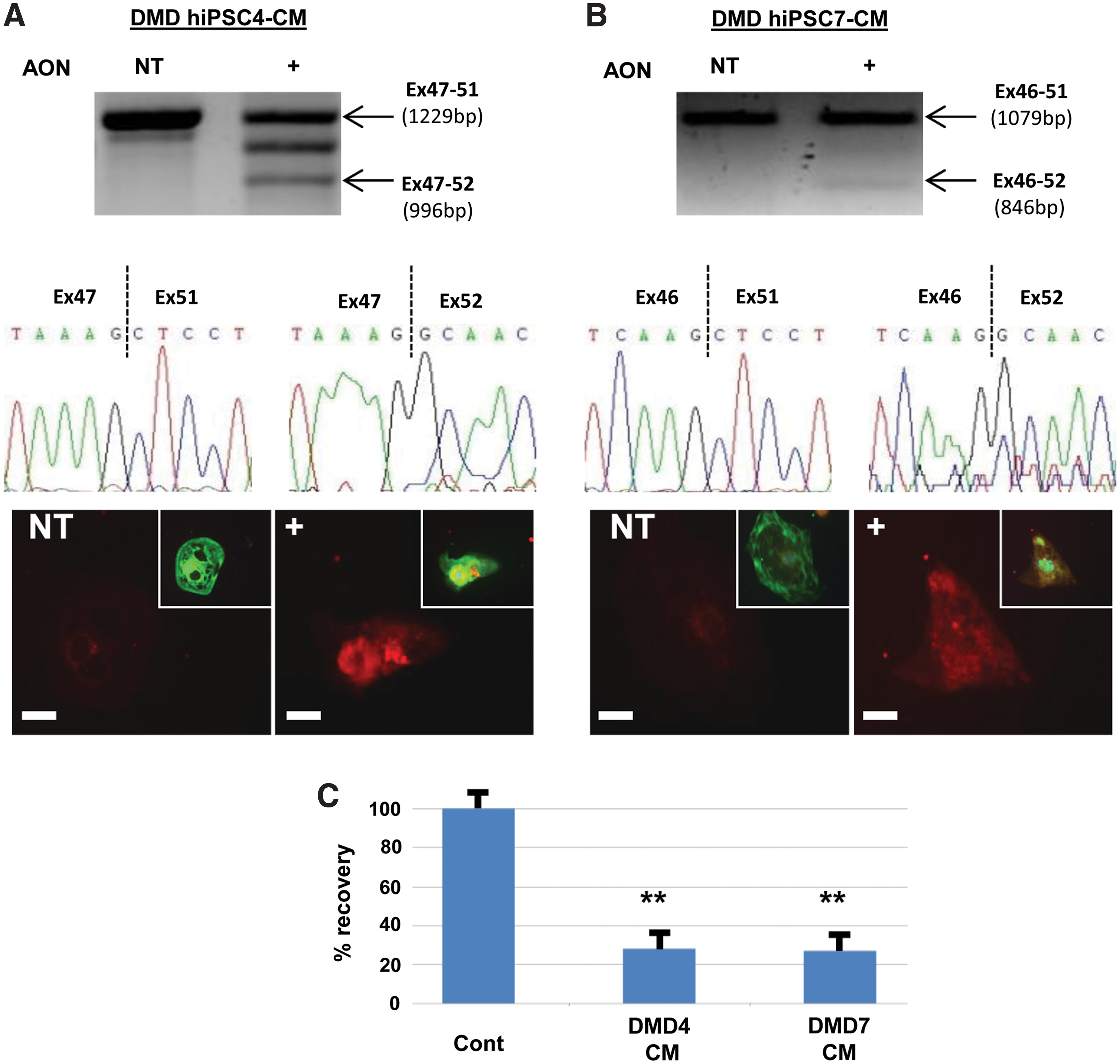
Exon skipping restores dystrophin expression in DMD hiPSC.
Since mini dystrophin constructs are being evaluated as potential therapies in the skeletal muscle of DMD patients, we sought to evaluate whether a 4.5 kb construct could restore expression in DMD hiPSC cardiomyocytes. We selected DMD hiPSC16 that harbors a stop mutation in exon 70, which is located outside the redundant central rod domain and hence exon skipping is unlikely to render a functional protein. This mutation disrupts all three dystrophin isoforms expressed in cardiomyocytes derived from hPSCs. In addition, we tested the minigene in DMD hiPSC19. The dinucleotide deletion in this line lies in region 1 (exon 35) and hence only disrupts the longest isoforms expressed in hPSC-derived cardiomyocytes. It also is outside of the exon 45–55 mutation hotspot region that is an appealing target for exon skipping. Lentiviral delivery of a 4.5 kb dystrophin minigene to cardiomyocytes derived from a healthy control DMD hPSC and from DMD hiPSC16 resulted in strong reverse transcriptase (RT)-PCR signals after minigene-specific RT-PCR primers (Fig. 6B). At the protein level, cardiomyocytes were co-stained with cardiac troponin I and C-terminal dystrophin antibodies. This showed lentiviral transduction led to expression of the minigene in cardiomyocytes derived from DMD hiPSC16 and DMD hiPSC 19 at levels of 66% and 91%, respectively, of healthy, nontransduced control hPSC-cardiomyocytes (Fig. 6C, D). These levels were achieved because the cardiomyocyte transduction efficiency was ∼55% and these cells expressed dystrophin at up to 1.6-fold relative to healthy control cardiomyocytes (data not shown).
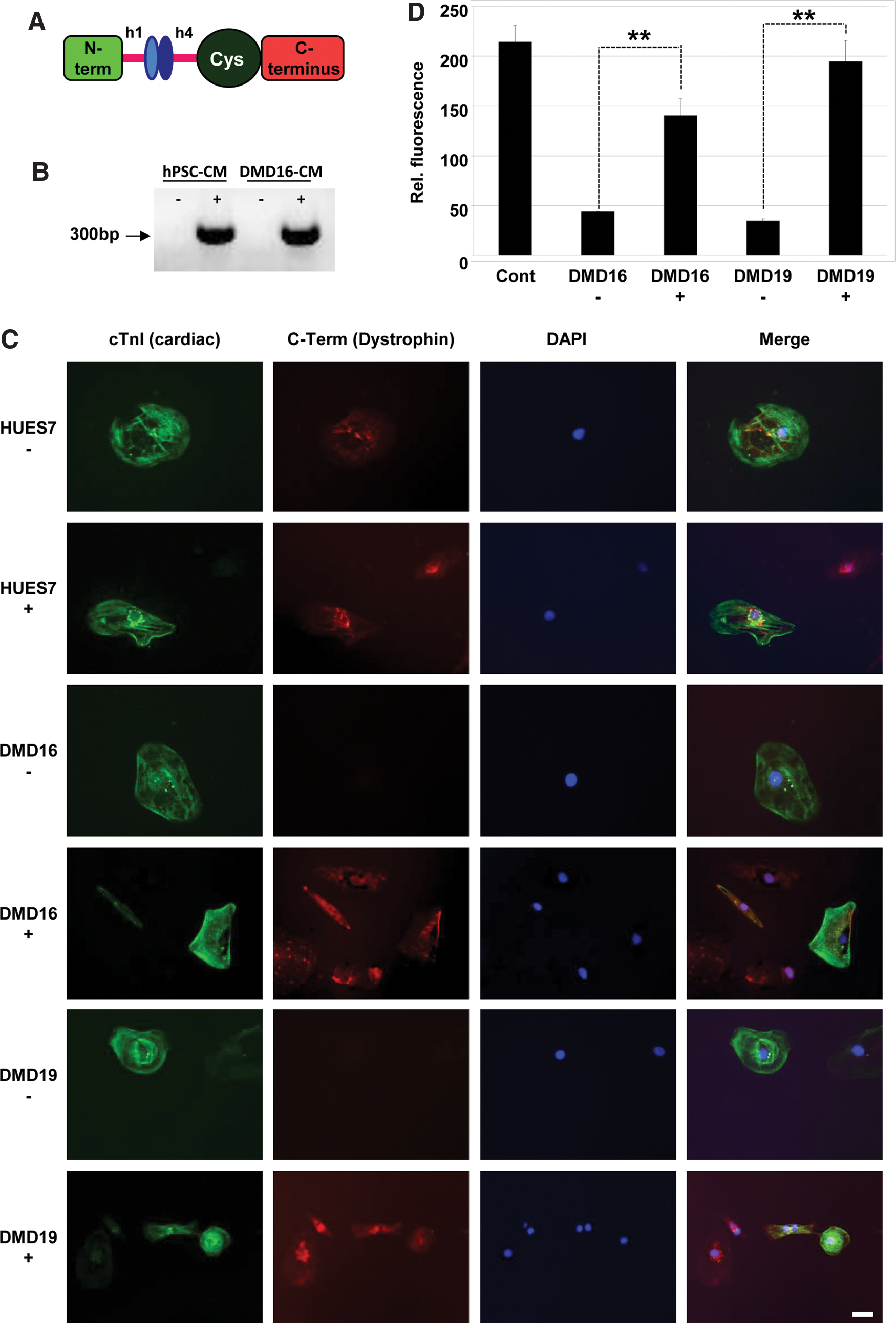
Dystrophin minigene expression in DMD hiPSC.
Thus, the DMD hiPSC model was successfully used to determine whether gene transfer and exon skipping strategies could restore dystrophin expression in cardiomyocytes, thereby providing a useful in vitro test platform.
Discussion
Our work shows that biopsies from DMD patients can be reprogrammed to hiPSC and differentiated into functional cardiomyocytes, which can be used as a model human system to test whether genetic-based therapeutics can restore dystrophin expression. The ease of transfecting hiPSC cardiomyocytes with 2OMePS AONs will be useful in defining which target sequences cause successful exon skipping in the heart, and whether this differs from cultured skeletal muscle cells. It also allows testing the ability of different mini- and micro-dystrophin constructs to produce protein in heart cells. Here, we tested delivery of a dystrophin minigene using recombinant lentivirus, although the system could be used to evaluate any viral or nonviral method. Clinical trials (see clinicaltrials.gov) are currently being initiated or are underway using lentiviral-mediated delivery of transgenes for esophageal cancer, skin disorders (e.g., Netherton Syndrome), and age-related macular degeneration, thereby providing therapeutic premise for the work presented in this article. Interestingly, cardiac problems are frequent in Becker patients and sometimes are the predominant symptom [26]. Thus, it is possible that while Becker-like dystrophins can improve skeletal muscle quality and function, they are less functional in the heart. Testing delivery in human cardiomyocytes and myoblasts in parallel will be important because cardiomyocytes have higher levels of RNA and higher RNA processivity, coupled with turnover of RNA. Thus, therapeutics discounted for low efficacy in myoblasts may be useful in cardiomyocytes.
Defining suitable target sequences will also allow their incorporation into other oligonucleotide-based strategies that are more difficult or expensive to test in culture. For example, it is challenging to deliver PMOs to cells in culture on account of the neutral charge of these skipping drugs [24]. The hiPSC-cardiomyocyte could also be used to test delivery and efficacy of pPMOs. Delivery of AONs expressed from viral vectors (e.g., adeno-associated virus [27] or lentivirus [28]) has been achieved but this strategy is costly and it is time consuming to screen large numbers of target sequences. Similarly, the system could be used to test the efficiency of uptake of new AON chemistries or methods of uptake, such as the use of cell-penetrating peptides [29,30]. It is notable that even when methods deliver limited quantities of skipping agents, dystrophin expression can be restored. For example, it has been shown that low skipping levels restored dystrophin to ∼75% of normal in cultured muscle cells [31].
Another advantage of the in vitro cardiomyocyte platform is that it will enable drug-induced cardiotoxic effects to be investigated. Cardiomyocytes from hESC and hiPSC have been validated in detection of drug-induced electrical disturbances, structural changes (e.g., hypertrophy), and reduced survival [14,32,33], providing opportunities to use these cells in toxicity testing of genetic-based therapies. The next stage will be to refine the DMD hiPSC-cardiomyocyte model so that in vitro function can be correlated with genotype and clinical pathophysiology. This has been achieved for conditions such as long QT syndrome [14], where electrical abnormalities in the patient's heart are reflected in the derived hiPSC-cardiomyocytes in culture. Such systems are now being used as a platform to test efficacy of experimental therapeutics. Making similar correlations for more subtle genotype–phenotype correlations that affect cell structure or integrity will be more challenging but also holds considerable reward once achieved.
Footnotes
Acknowledgments
The Medical Research Council, Biotechnology and Biological Sciences Research Council, British Heart Foundation, and the University of Nottingham funded this study. Fibroblasts were received from the Medical Research Council (MRC) Centre for Neuromuscular Diseases BioBank, Newcastle, which is a member of the EuroBioBank Network (
Author Disclosure Statement
The authors declare no conflict of interest. No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.