
Abstract
Mesenchymal stem (or stromal) cells (MSCs) are nonhematopoietic progenitor cells that can be obtained from bone marrow aspirates or adipose tissue, expanded and genetically modified in vitro, and then used for cancer therapeutic strategies in vivo. Here, we review available data regarding the application of MSC-based tumor-targeted therapy in gastrointestinal cancer, provide an overview of the general history of MSC-based gene therapy in cancer research, and discuss potential problems associated with the utility of MSC-based therapy such as biosafety, immunoprivilege, transfection methods, and distribution in the host.
Background
T
Researchers have recently made use of MSCs as delivery vehicles for tumor-targeted gene therapy, in part, due to their accessibility for genetic modification in vitro and their ability to be cultured and expanded in vitro [5]. The cells are easily obtained from a simple bone marrow aspirate and can be readily expanded [6]. Their extraordinary high proliferative capacity is thought to contribute to the in vivo maintenance of both tumor stroma and connective tissue in organs remote from the bone marrow [7]. MSCs successfully engraft into tissues under conditions of increased cell turnover, for example, those triggered by a tissue damage or neoplastic growth. They have the ability to efficiently home to sites of tissue injury, including tumor environments [8,9]. The exact mechanism governing this recruitment is not well understood. MSCs are thought to show a strong tropism for tumors, because the body sees the tumor environment as the equivalent of a chronic wound–for example, “the wound that never heals” [10 –13]. Furthermore, MSCs inhibit T-cell proliferation [14,15], induce T-cell apoptosis [16], alter migratory property of T-cells [17], and are resistant to natural killer cell-mediated cytolysis [18]. These observations provide an explanation for the immunoprivileged nature of MSCs. Each of these attributes can be seen to contribute to the potential application of MSCs for cell-based delivery of therapeutic genes to solid tumors.
Genetically Engineered MSCs in Nongastrointestinal Cancer Therapy
Molecules that physiologically control cell proliferation are often produced locally in tissues, but are rapidly turned over when they enter the peripheral circulation [e.g., transforming growth factor (TGF)-β, tumor necrosis factor-α, interleukin (IL)-2, and interferon (INF)- β] [19 –21]. The broad application of these biologic agents in cancer therapy is generally limited by their short biologic half-life or excessive toxicity [22]. For effective antiproliferative therapy, the biological concentration of these or other therapeutic agents required to achieve a therapeutic effect can often be substantially higher than serum levels achievable after systemic administration at the maximally tolerated dose [23 –25]. Similar issues arise when one considers general questions of regional-versus-systemic therapy using more focused biologic approaches, for example, the use of suicide gene therapy [26].
An early application of MSCs as vehicles for cancer therapy was described by Studeny et al. [27]. The authors transfected hMSCs with the interferon IFN-β, which were used to treat melanoma xenografts in mice. Injection of the transfected MSCs into the peripheral circulation lead to reduced tumor growth and prolonged survival of tumor-bearing mice. Subsequently, MSCs from different sources, including human bone marrow-derived MSCs [28 –31], human adipose tissue-derived MSCs (hAT-MSCs) [32,33], human umbilical cord blood-derived mesenchymal stem cells [34], mouse bone marrow-derived MSCs (mMSCs) [35 –38], and rat MSCs [39 –42], have been evaluated as vehicles for tumor therapy. The expression of diverse therapeutic genes, including IFN-β [27,28,30], TRAIL [29,32,34], PEDF [33], IL-12 [35], CX3CL1 [36], VEGFR-1 [38], iNOS [42], and HSV-Tk [37,39 –41], has been engineered into MSCs to allow a targeted release of these agents in models of melanoma [27,28], breast cancer [29,35], Lewis lung carcinoma [36], gliomas [30,34,37,39], glioblastoma [40,41], cervical cancer [32], prostate cancer [33], and fibrosarcoma [42]. In each of these tumor models, treatment showed efficacy in the inhibition of local tumor growth, suppression of metastasis, or prolongation of animal survival (Table 1).
hBM-MSC, human bone marrow-derived mesenchymal stem cell; hAT-MSC, human adipose tissue-derived mesenchymal stem cell; hUCB-MSC, human umbilical cord blood-derived mesenchymal stem cell; mMSC, mouse bone marrow-derived mesenchymal stem cell; rMSC, rat bone marrow-derived mesenchymal stem cell.
Application of MSCs as Tumor-Targeted Therapy Vehicle
Biosafety of MSCs
Houghton et al. [43] suggested that MSCs can potentially act as cancer progenitors in a chronic Helicobacter-infected gastric cancer model. The authors suggested that bone marrow-derived cells can home to and repopulate the gastric mucosa and contribute over time to metaplasia, dysplasia, and cancer. Tolar [44] had shown that mice implanted with high passage-number MSCs can form sarcomas in situ. Importantly, these mice had been lethally irradiated before the MSC administration. Numerous reports have also shown that long-term cultures of murine MSCs can undergo spontaneous malignant transformation [45 –47]. These issues have not been reported in the thousands of patients who have undergone MSC engraftment in the context of tissue-repair studies or for treatment of graft-versus-host disease [48].
Importantly, when using engineered MSCs, it is possible to design transgenes that allow the elimination of all adoptively transferred MSCs in the context of therapy. This can occur in the course of treatment (e.g., with the use of a suicide gene). We have found that even a very low expression of the herpes simplex virus–thymidine kinase (HSV-Tk) transgene seen in MSCs that migrate to nontumor tissue is still sufficient to make the cells sensitive to treatment with gancyclovir, but not strong enough to damage the resident tissues. In addition, as an additional level of security, it is also possible to add an additional transgene for wide-scale elimination of all adoptively transferred MSCs in patients.
MSCs are immune privileged
MSCs are an interesting therapeutic tool, as they are largely immune privileged. These cells lack the expression of MHC class II, show low expression of MHC class I, and lack expression of CD40, CD80, and CD86 [12]. Plastic-adherent adipose-derived stem cells (ASCs) beyond passage P1 have been shown to be unable to induce a response from T cells. Consistent with these findings, late-passage ASCs can inhibit mixed lymphocyte reactions (MLRs) [49]. These findings suggest that therapies using allogeneic MSCs may also be immune suppressive, and clearly this is a relevant issue that must be addressed in the context of tumor therapy.
Transfection methods
Studeny et al.[27] used an adenovirus as a transfection vector for engineering antitumor MSCs. Kucerova et al. [11] subsequently applied retrovirus-transfected cytosine deaminase (CD) into hAT-MSCs against colon cancer. Adenoviral vector expression is transient and produces a host-immune response, whereas retroviruses have been linked to insertional mutagenesis. Lentiviral vectors, which were first utilized by Loebinger [29] in hMSCs transfected with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), were described to be less likely to cause vector-induced expression of insertion-proximal genes, as the long terminal repeats can be modified to remove their promoter activity. Lentiviral vectors are also capable of stably transducing both dividing and quiescent cells [50].
OriP/Epstein-Barr virus (EBV) nuclear antigen (EBNA)-based episome plasmids have been shown to be retained in cells after cell division. Hybridized EBNA-1 elements of the EBV vector with herpes simplex virus type-1 (HSV-1) have shown high vector levels and prolonged transgene expression. EBNA-based episome may represent an alternative nonintegrating platform for anticancer-engineered MSC therapy [51].
Transposon-based gene vectors are an emerging technology for gene therapy of MSCs. The 3 most used transposon-based vectors are the Sleeping Beauty (SB), piggyback, and Tol2 systems. These vectors provide long-term transgene expression in human cells with minimal signs of silencing. In addition, they have the added advantage that they are nonviral delivery systems. They are relatively simple to use; they can be used to express rather large and complex transgenes, are low-cost, and have low-immunogenicity. To date, the SB system has been the most widely applied. It is an efficient system that can integrate transposon DNA in diverse cell types, including human hematopoietic stem/progenitor cells [52,53].
Distribution of adoptively applied MSCs in the host
In healthy mice, exogenously applied MSCs quickly show up in the lung. MSCs that are cultured in vitro are generally rather large. After injection, their size and the small dimensions of the lung vessels cause their temporary residency in the lung. Over time, the cells leave the lung and move to other organ systems. In normal mice, adoptively transferred MSCs show migration to spleen, thymus, lymph nodes, mucosa of the small intestine, skin, and salivary glands [54].
The complement of chemokine receptors found on MSCs helps dictate their homing in normal and disease settings [54]. Human and mouse MSCs grown in culture have been shown to functionally express the chemokine receptors CCR1, CCR2, CCR3, CCR4, CCR7, CCR8, CCR9, CCR10, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, and CXCR6 [55 –58]. These receptors are thought to be involved in the recruitment of MSCs to skin, gut mucosa, and salivary glands [54]. These receptors may also be linked to the recruitment of MSCs to the tissue damage associated with tumor stroma or response to wound healing.
In addition, tissue inflammation or damage also leads to the production of endocrine signals that may also help attract MSCs. As previously discussed, tumor microenvironments are thought to show histologic similarities to those seen in normal wound healing [10]. In a similar fashion, tumors efficiently attract MSCs, recruited from both the bone marrow and adoptively transferred sources. MSCs integrate into the tumor stroma where they help foster tumor development and progression.
These general phenomena have been demonstrated by diverse studies. For example, Studeny et al. [27] reported that after intravenous injection of INF-β-transfected hMSCs in a metastatic melanoma lung model, MSCs were randomly distributed in the lung parenchyma and in the melanoma metastases by 24 h after injection. Interestingly, 8 days later, the MSCs were preferentially found at the periphery of the metastases, and after 60 days, numerous MSCs were diffusely present in the lung metastases. In an orthotopic liver cancer model, Gao [59] analyzed fluorescent signals 7 days after intravenous injection of CM-Dil-labeled hMSCs. The cells localized mainly around the tumor, with rare cells were found in the lung. Similarly, in an orthotopic pancreatic cancer model, Beckermann et al. [60] showed that 3 days after MSC injection, most of the adoptively applied cells were recruited to the tumor, and only few MSCs were found in the liver, lung, and spleen.
It has recently been shown that due to its dual role as reporter and therapeutic gene, the sodium iodide symporter (NIS) allows noninvasive imaging of functional NIS expression by 123I-scintigraphy or 124I-PET imaging before the application of a therapeutic dose of 131I. MSCs transfected with NIS cDNA were injected in a hepatocellular carcinoma (HCC) xenograft mouse model. NIS allowed monitoring the distribution of NIS-MSCs by 123I-scintigraphy and 124I-PET imaging after intravenous injection of NIS-MSCs. Systemic MSC-mediated NIS gene delivery followed by 131I application resulted in a significant delay in tumor growth. The results show tumor-specific accumulation and therapeutic efficacy of radioiodine after MSC-mediated NIS gene delivery in HCC tumors [61]. We have also injected MSCs labeled with supermagnetic iron oxide (SPIO) into mice with a growing orthotopic HCC. Their recruitment and distribution were then monitored under an MR image. SPIO-labeled MSCs were detected after 36 h in the tumors of mice. No signal was detected in other organs, which was confirmed by immunohistochemistry [62].
Routines of MSC application
In experimental settings, the most common means of administration of MSCs is via the intravenous or intratumoral route. Ram et al. [63] performed direct intratumoral injection of murine HSV-Tk vector-producing cells in recurrent malignant brain tumors of patients. However, the therapeutic response suggested a poor clinical utility. It is possible that the normal physical processes associated with MSC recruitment from the peripheral circulation may impart important differentiative signals. For example, systemically applied autologous hMSCs in patients suffering from cerebral infarction or multiple-system atrophy resulted in delayed disease progress and were safe for those patients at a 1-year follow-up time [64,65]. A similar phenomenon may help explain the disparent results outlined in Table 2. Although intratumoral injection may be feasible in some clinical applications, such as in the context of percutaneous ethanol injection or radiofrequency ablation in liver cancer, the imprinting MSCs receive during their passage through the peripheral circulation may be important to their eventual recruitment and differentiation in a tumor setting. For most experimental gastrointestinal tumors, intravenous application of engineered MSCs has generally shown better efficacy.
Lin-CD44hiSca1-cKit+CD34- MSC.
transfected without IFN-β.
immortalized by hTERT.
CD, cytosine deaminase; GCV, ganciclovir; IFN, interferon; GFP, green fluorescent protein; PEDF, pigment epithelium-derived factor; RFP, red fluorescent protein.
Numbers of MSCs administered
The number of MSCs required to generate a therapeutic effect remains an open variable. Known is that the application route of MSCs, the kinetics of MSCs in tumors in situ or in circulation, the origin of MSCs, and the specific type of therapeutic gene expressed need to be taken into consideration.
In the various experimental settings, the number of MSCs used to achieve a therapeutic effect has generally been comparable to the number of tumor cells used to establish the tumor model. Although, the number of tumor cells that survive injection to seed tumor growth can be rather small [66,67]. Importantly, even at the highest cell dose examined (2–2.5×108 AT-MSCs/kg in mice, 1.2×106 AT-MSCs in rat, or 4×108 AT-MSCs in humans) [68,69], the application of cells appears to be safe.
Dual Effects of MSCs on Gastrointestinal Cancers
MSCs are recently under investigation for hepatic regeneration and hepatocyte differentiation [70]. Regenerative medicine holds much promise for the development of cell-based therapies based on the MSC biology [71,72]. It also suggests a complex biology of MSCs that should be addressed in future studies.
As shown in Table 1, in nongastrointestinal cancers, MSCs have demonstrated comparable effects regarding tumor growth, metastasis, and animal survival. Control, or nontherapeutic, MSCs are also recruited to the tumor site where they can function as stroma cells to support tumor development. However, in gastrointestinal cancers, the biology appears more complicated, as conflicting data exist regarding the biology of MSCs in these tumor settings. After subcutaneous co-injection of liver cancer cells and hMSCs transfected with the human telomerase reverse transcriptase (hTERT) gene, Qiao et al. [73] showed that the engineered hMSCs could inhibit tumor growth by downregulation of NF-κB- or Wnt-signaling pathways [74]. In contrast, our group has found that systemically applied MSCs can strongly promote tumor growth in orthotopic pancreatic [67,75] and HCC models [62]. In studies where MSCs were engineered to express human pigment epithelium-derived factor (PEDF) [59] or HSV-Tk [62,67,75], it has been shown a significant suppression of pancreatic and hepatic tumor growth (Table 2).
Similar experimental settings have shown apparently disparate results concerning the biological effects of MSCs. Li et al. [76] reported that hMSCs can enhance tumor growth in vivo in an HCC s.c. model, whereas the MSCs were found to inhibit the invasion and metastasis of the same cell type in in-vitro settings. Interestingly, expression levels of TGFβ1 by MSCs were decreased in both the in vitro and in vivo experiments.
MSCs appear to have a complex biology in other gastrointestinal cancers, including esophageal cancer, gastric cancer, and pancreatic carcinoma. Li et al. [77] applied hMSCs together with esophageal cancer cells subcutaneously in nude mice. The authors showed that hMSCs could promote tumor growth with increased tumor vessel formation in vivo. Interestingly, the MSCs were found to inhibit the proliferation and invasion of tumor cells in vitro. These effects were again associated with a general downregulation of canonical Wnt signalling [73,78]. In a gastric cancer xenograft mouse model, You et al. [79] injected hMSCs transfected with the suicide gene cytosine deaminase (CD), which was followed by treatment with the prodrug 5-fluorouracil (5-FU). This resulted in a pronounced inhibition of tumor growth. In a chronic Helicobacter felis–induced gastric dysplasia mouse model, Wang [80] applied murine bone marrow-derived Lin-CD44hiSca1-cKit+CD34-MSCs via tail vein injection. Surprisingly, these MSCs were found to reduce tumor progression to low-grade gastric dysplasia, and correlated with reduced gastric IL-17F, IL-22, and ROR-γt gene expression. Kidd et al. [81] showed that hMSCs with or without transfected IFN-β were both found to suppress tumor growth in the same orthotopic pancreatic cancer mouse model. This is in contrast to the results of our studies in an orthotopic pancreatic cancer model [62,67], where control MSCs were found to strongly promote primary tumor growth and to increase metastases. However, suicide gene (HSV-Tk)-transfected MSCs were shown to substantially inhibit local pancreatic tumor growth and the incidence of metastases [67] (Table 2).
Additional Examples of Genetically Engineered MSCs in Gastrointestinal Cancer Therapy
As we have seen, apparently, contradictory reports of the biology of MSCs have been described in gastrointestinal cancers (Table 2). There are additional examples showing coincidence with the observation of MSCs promoting tumor growth and therapeutic transfected MSCs inhibiting tumor growth. Shinagawa [82] intravenously injected hMSCs in an orthotopic colon cancer model, which resulted in an enhancement of tumor growth and metastases. Using CD-transfected hAT-MSCs in a colon adenocarcinoma xenograft model, Kucerova et al. [11] could show tumor growth inhibition. Moreover, Chen [83] and Hu [38] transfected mMSCs with IL-12 and VEGF-1 and then successfully demonstrated prevention of colon cancer carcinogenesis in a mouse model, reduction of lung metastasis, and a prolongation of lifespan. Studeny published the first 2 reports describing hMSC-based gene therapy in tumor models [27,28]. Wolf [84] argued that the selective homing of systemically injected human MSCs in this model might be too artificial to be relevant to the in vivo clinical situation. To address this, the authors used a complete syngenic murine model as an experimental system. In addition to MSC engraftment into syngenic tumors, the authors also observed exogenously applied MSCs at additional tissue sites, including the spleen, liver, and normal lung [84]. The authors suggested that human tumors may selectively attract human MSCs by secretion of human-specific chemoattractants. Our group has previously shown [67] that green fluorescent protein (GFP)-transfected mMSCs in an orthotopic murine pancreatic cancer model are effectively recruited to the tumor, but some signals were also found in the spleen, lymph nodes, thymus, skin, and gut. More recent studies using imaging in xenogenic models support the powerful effect that solid tumors have on the recruitment of adoptively transferred MSCs, whether syngenic or xenogenic, to the tumor site [62].
The potential recruitment of adoptively transferred MSCs to nontumor tissue environments with associated side effects is a potential concern for the general adaption of this technology for the treatment of cancer. One approach is to attempt to specifically direct the expression of the transgene only to tissue-specific environments, for example, by using specific gene promoters. Studeny [28] made an early referral to the use of specific promoters. The authors transfected IFN-β into hMSCs using an adenoviral vector system, and—as expected—found that the inhibition of tumor cell growth by MSC-IFN-β cells was not permanent. Adenoviral vectors generally lack a sustained effect, as adenoviral transgenes do not integrate into the genomes of transduced cells, and the transgene copy number per cell declines, as the virally engineered MSCs proliferate in tumors. Later, Loebinger [29] made use of the inducible Tetracycline-on system to activate MSCs transfected with TRAIL to treat different cancers. The Tet-on system allowed TRAIL effector and GFP reporter gene expression to be induced under the control of a tetracycline promoter in MSCs using a lentiviral vector system.
Our group has approached this question from a different perspective through the use of tissue-specific promoters. This concept makes use of the differentiative capacity of MSCs after their recruitment into tumor microenvironments, resulting in a more restrictive expression of a therapeutic transgene only in a specific tissue context (activation of the transgene by tissue-specific signals), thus potentially sparring nontumor tissues from therapeutic damage.
First, we made use of the observations of Karnoub et al. [85], who studied the role of MSCs in a xenograft model of breast cancer. The authors demonstrated that MSCs are actively recruited into a tumor-associated stroma. Once there, the MSCs actively secrete the chemokine CCL5. We evaluated the use of the CCL5 promoter to drive the expression of the suicide transgene HSV-Tk (accompanied with ganciclovir [GCV]) in engineered MSCs in a syngenic model of pancreatic cancer [67]. After verifying the induction of CCL5 by MSCs in the context of pancreatic cancer using reporter genes, the promoter was then used to induce the expression of HSV-Tk. After treatment with GCV, strong inhibition of tumor growth could be demonstrated with this selective targeting of the pancreatic cancer tumor stroma. Importantly, this treatment also significantly reduced metastases in this model. In a second set of experiments, expression of HSV-Tk was driven by the Tie2 enhancer/promoter. The idea was to drive the transgene expression in engineered MSCs, only when a subgroup of tumor infiltrating MSCs differentiate to endothelial-associated cells in the context of tumor angiogenesis. The Tie2-targeting strategy also effectively inhibited primary tumor growth of experimental pancreatic cancer [75].
In each of these settings, this refined targeted therapy was achieved only when the engineered MSCs infiltrate the tumor and undergo differentiation and consecutive promoter-driven activation of the transgene. We refer to this as a Trojan Horse MSC therapy. Even if engineered MSCs reach other organs, the effect of the transgenes will be limited to the tumor site, as they are under the control of promoters that are not active in other tissue settings (Fig. 1).
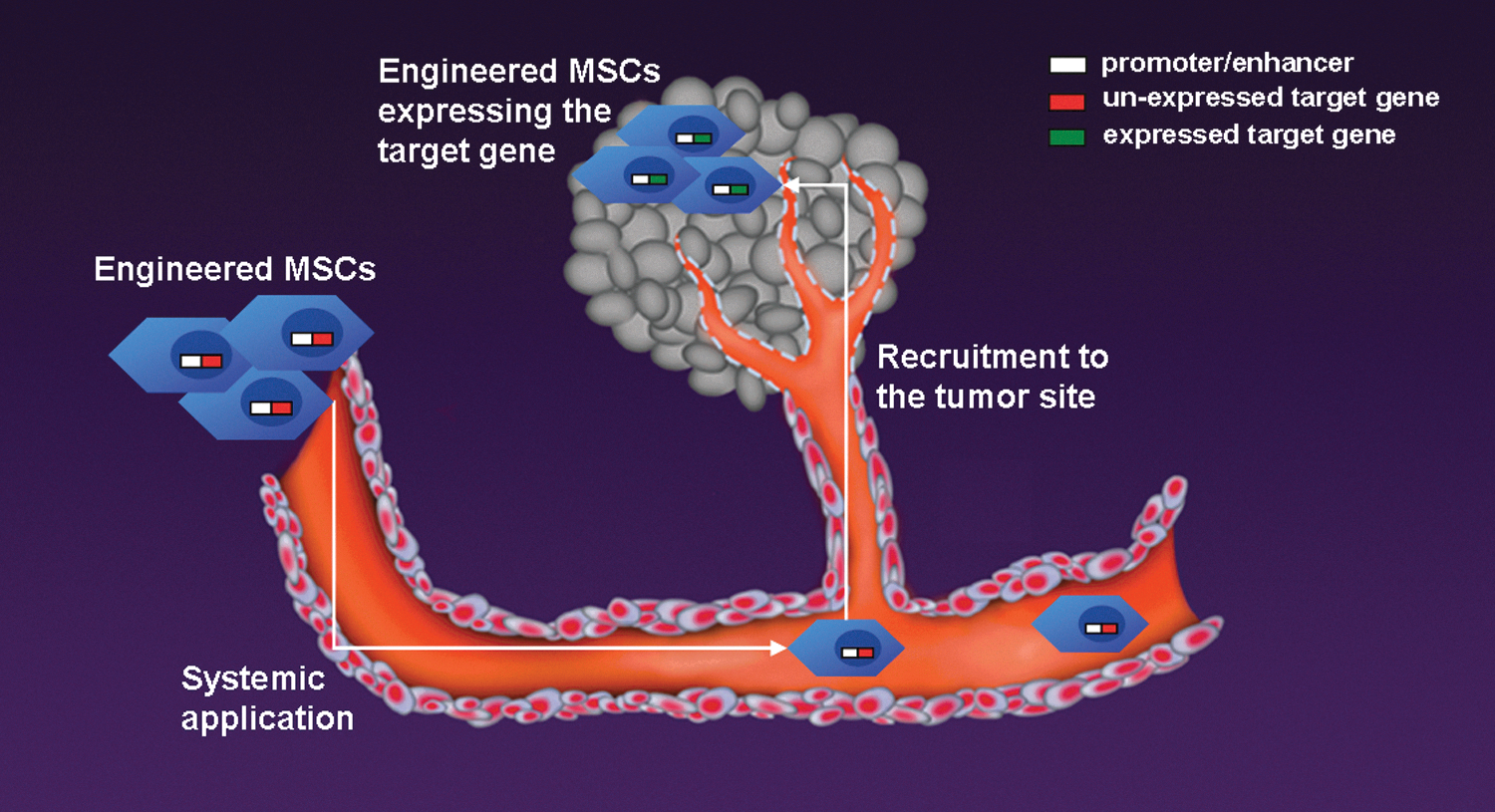
Schematic view of engineered mesenchymal stem cells (MSCs) as targeted tumor therapy (“Trojan Horse” MSC therapy). Therapeutic genes under the control of a promoter/enhancer are transfected into MSCs. After systemic application, engineered MSCs home to the tumor environment where they undergo differentiation and consecutive promoter-driven activation of the transgene.
Conclusions
During the past decade, a remarkable progress has been made in the area of engineered MSC-based therapy, especially in the context of gastrointestinal tumors. Although there is still controversy about the exact function of MSCs in these tumor settings, a series of common characteristics of these cells are now largely accepted by researchers. MSCs have numerous advantages as tumor-target-therapy vehicles, as they are easy to isolate and expand in vitro, can be efficiently transfected with various vector systems, migrate to and integrate into tumor environments after systemic delivery, and have immunoprivileged properties.
Additional work needs to be done before clinical trials are initiated. For instance, as the frequency of MSCs in human bone marrow is low (0.01%–0.001%), although they can be isolated from adipose tissue, there is still a significant requirement for extensive ex vivo expansion before use. Further understanding of the biological properties of MSCs and their kinetics in vivo will help foster the translation of this approach to the clinic.
Footnotes
Author Disclosure Statement
Ralf Huss works for a company with the goal to commercialize MSC-based therapy. The remaining authors declare no conflict of interest.