
Abstract
Bacteriophage recombinases can target specific loci in human embryonic stem cells (hESCs) at high efficiency, allowing for long-term expression of transgenes. In the present work, we describe a retargeting system where we used phiC31 integrase to target a plasmid to a pseudo-
Introduction
H
Several viral and nonviral techniques have been employed to deliver genetic elements to hESCs [1 –5]. Lentiviral particles, for instance, have been a popular vehicle for gene delivery to hESCs due to their high infectivity and efficient stable integration of expression constructs into the genome [6 –9]. While this platform is arguably the most efficient of current techniques at delivering DNA to hESCs, its use is restricted in some cases due to limited DNA payload capacity (6–8 kb), the random nature of genetic insertion, and limited control of copy number of the inserted genetic element.
Regardless of the mode of gene delivery for a particular experiment, expression of the transgene is significantly affected by the surrounding activity of its genomic insertion site. Confounding this “position effect” is the fact that as hESCs differentiate, they go through chromatin remodeling leading to silencing of some loci and potential activation of others [10 –13]. Expression cassettes randomly inserted into the hESC genome (via viral or other mode) are subject to seemingly random locus-specific silencing during differentiation, potentially leading to heterogeneous expression patterns in the differentiated cell population. This heterogeneity is compounded by the lack of control of copy number and the necessity of performing multiple delivery events if one desires expression of multiple genes. All these issues taken together can lead to unpredictability and cell-to-cell variability.
We describe here the development of a system, which is intended to mitigate many of these issues by creation of a “platform” hESC line where large, complex genetic elements can be stably introduced into cells at a defined chromosomal locus in single copy. The target locus has been validated for high retargeting efficiency, stability through multiple passages, high transcriptional activity in hESC, and resistance to silencing upon differentiation.
The system uses 2 families of integrase enzymes first to assemble the targeting plasmid (lambda integrase in Multisite Gateway) [14
–18] and the second to create a “platform” site in the hESC genome that can be targeted efficiently. Both integrases, phiC31 and R4, from the
In the present work, we used 2 hESC lines, BG01V and H9, and phiC31 integrase to place a native R4 attP site (a platform for future genetic engineering) to a transcriptionally active and stable locus in the hESC genome, then to efficiently “retarget” that site using R4 integrase with expression constructs. We describe construction of this “R4 platform line” followed by mapping of the target site locus and retargeting the site with a constitutively expressing EmGFP construct. We use this retargeted R4 hESC line to assess transcriptional activity of the site, retargeting efficiency, stability of expression through multiple passage, and resistance to locus silencing upon differentiation of the reporter line to multiple lineages.
Materials and Methods
Vector construction
The plasmid pJTI/Zeo was cloned as follows. The plasmid pB2H1-Z1 was generated by cloning in a filled-in
The plasmid pER4B-DEST (pJTI R4DEST; Life Technologies, Carlsbad, CA) was cloned as follows. The R4
Cell culture
H9 (46, XX) and BG01V hESC line (48, XY, +12, +17) were maintained as described [26,27]. In brief, cells of both cell lines were cultured on a layer of mitomycin C (Sigma, St. Louis, MO)-inactivated mouse embryonic fibroblast cells (MitC-MEF) in hESC medium containing DMEM-F12, 20% knockout serum replacement, 1% nonessential amino acid, 55 µM 2-mercaptoethanol, 2 mM
Transfection, retargeting, clone selection, and maintenance
To generate the targeting platform line, electroporation was carried out as previously described [23]. In brief, 6 million cells were harvested using Accutase (Sigma) or TrypLE (Life Technologies) and resuspended in 800 µL of OptiPro™ SFM (Life Technologies) with R4 platform vector and phiC31 integrase at a mass ratio of 1:1 to 1:3 (total plasmid mass = 30 µg). These cells were placed in an electroporation cuvette with a gap of 0.4 cm. Cells were electroporated with a pulse of 200 V at 10 ms, 2 pulses using the ECM830 electroporator (BTX). Electroporated cells were plated on MEF feeders and allowed to recover for 48–72 h before selection was started with hygromycin (10 µg/mL; Life Technologies). After 14–21 days of selection, individual drug-resistant clones were manually picked and expanded for further analysis and retargeting.
To retarget the platform lines, “R4” cells were harvested using TrypLE and 1 × 106 cells were electroporated using a microporator (Digital Bio) at 850 V, 30 ms with 10 µg of a retargeting construct containing a promoter-GFP cassette (in the present work, either EF1α or Oct4 promoter is used, ie, plasmids pER4B-EG and pER4B-hOG), which when site-specifically integrated into the platform line by R4 integrase activates expression of the Zeocin-resistance gene expression. Ten micrograms of codon-optimized pCMV-R4 integrase expression plasmid was co-transfected along with the retargeting vectors. Transfected cells were seeded on hygromycin-resistant MEF layers (Millipore, Billerica, MA) and allowed to recover for 48 to 72 h. R4 integration-mediated retargeted clones were selected using 2.5 µg/mL Zeocin (Life Technologies). Colonies were picked as described earlier.
Southern blot verification and plasmid rescue
Genomic DNA from individual clones was isolated using either the ChargeSwitch gDNA Mini Tissue Kit (Life Technologies) or DNAzol Reagent (Life Technologies). Plasmid rescues were done as described [23]. Southern blots were performed as follows. Genomic DNA (20 µg) from parent clones was restricted with
The presence of a retargeting event was checked by amplification of genomic DNA isolated from individual clones. The primers used for detecting these events were 5′-GAGCATGCATCTAGTCCAGTGTGG-3′ and 5′-CATGG TTTAGTTCCTCACCTTGTCG-3′. The absence of genomically integrated phiC31 integrase or R4 integrase expression plasmids was determined by PCR amplification of genomic DNA. Primary PCR was followed by a secondary amplification with nested primers. The primary PCR primers used for phiC31 integrase were 5′-AGCGTAGCGCC AACGAAGA-3′ and 5′-TGAATGGCGGCTTGACTGC-3′, and secondary primers were 5′-GACGGGGGCCGGTTCAGG-3′ and 5′-CGCCTTCCCGCCGACGTAC-3′. The primers used for R4 integrase were 5′-GTCGGGAGCTCTGCAAGAGC-3′ and 5′-CCGCCAGCGGCCATAG-3′ for the primary PCR and 5′-GGACAATGACCTTTCCGCGAC-3′ and 5′-AGTGTA AGGCTTTCCGGCTCTAGC-3′ for the secondary reaction. Plasmid DNA was used as a positive control to ensure that even a single copy of the integrated plasmid would have resulted in amplification of a product.
Random differentiation of hESCs and directed neural differentiation of retargeted clones
Undifferentiated hESCs were harvested using collagenase to generate embryoid bodies (EBs) and were cultured for 4 days in suspension in differentiation medium containing DMEM-F12, 20% knockout serum replacement, 1% nonessential amino acid, 55 µM 2-mercaptoethanol, and 2 mM
Directed neural differentiation toward neural stem cells from hESC was performed using a modified protocol based on the procedure described in Benzing et al. [28]. In brief, hESCs were differentiated first as adherent cultures in NAA medium containing DMEM:F12, 1× N2, 200 mM ascorbic acid, 10 ng/mL bFGF for 6 to 9 days, then were cultured in suspension in NAA medium for another 8 to 12 days before they were seeded onto Geltrex-coated dishes for FACS analysis and immunocytochemistry or further differentiation. Neural stem and lineage precursor cell markers used to characterize the cells underwent the above neural differentiation include Sox1, Nestin, Musashi1, Pax6, A2B5, PSA-NCAM, and PDGFRα.
Flow cytometric analysis and immunocytochemistry
Retargeted clones, their parental non-engineered lines or differentiated cells, were harvested using TrypLE; cell debris were excluded from analysis by gating based on forward and side scatter. Data was collected using BD FACS CantoII Flow Cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
Immunocytochemistry was carried out as described [29]. In brief, undifferentiated hESC or differentiated cells were fixed with 2% paraformaldehyde and incubated with blocking buffer for 30 min. Primary antibodies were added to the cells and incubated at 4°C overnight followed by addition of appropriate secondary antibodies. The following primary antibodies were used: Oct4 (1:500; Abcam, Cambridge, MA), SSEA4 (1:500; Life Technologies, Carlsbad, CA), Tra1-60 (1:100; Millipore, Billerica, MA), Tra1-81 (1:100; Millipore), βIII Tubulin (Tuj1, 1:4,000; Sigma, St. Louis, MO), Nestin (1:500; BD Biosciences, San Jose, CA), Smooth muscle actin (1:200; Sigma), α-fetoprotein (1:500; Sigma), Sox1 (1:240; Millipore), Musashi1 (1:200; Millipore), A2B5 (1:200; Millipore), PDGFRα (1:200; BD), CNP (1:200; Sigma), GFAP (1:4,000; DAKO). Secondary antibodies were used at the following concentrations: Alexafluor 594- or 488-conjugated anti-mouse IgG (1:1,000) and Alexa 594- or 488-conjugated anti-rabbit IgG (1:1,000), allophycocyanin (1:1,000, all from Life Technologies). DAPI was used for counter nuclei staining. Images were captured using a Zeiss Axiovision microscope with Z-stack split view function and images processed using AdobePhotoshop CS.
Results
Strategy for site-specific targeting and retargeting
In this study, we exploited the ability of phiC31 integrase to target preferred sites at an easily detectable frequency by using it to place a target for the R4 integrase. A plasmid containing the R4
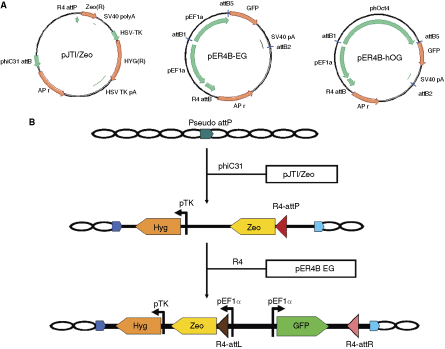
Retargeting strategy and plasmids used in this study. (
Generation and verification of pluripotent platform lines
Electroporation was used to generate the platform hESC lines using BG01V and H9 as described earlier. We mapped a set of 42 clones and identified the various chromosomal loci using plasmid rescue followed by data base alignment (BLAT) [23]. Of these, we were able to map the integration of the target plasmid in 23 clones that represented integration into 10 separate genomic loci, including five of the hESC-specific hot spots described earlier, chromosome 13q32, chromosome 6p25, chromosome 2q35, chromosome 10p12, chromosome 17q23, and chromosome 21q21 [23]. Mapped platform sites were evaluated for potential for silencing after differentiation by comparing expression profiles of the resident (if in an intron) or nearest neighbor genes in adult tissue using a microarray database. Loci were categorized according to balanced expression levels and used for further retargeting experiments. In a preliminary screen for retargeting using all clones targeted to different genomic loci, we were only able to retarget clones successfully that had the R4 attP target placed at the chromosome 13q32 hot spot. Based on the mapping data, 6 of the 23 mapped clones (∼25%) were targeted at chromosome 13q32, 3 in 1 orientation and 3 in the other. This chromosome 13 hot spot was therefore used for retargeting and further investigation.
After verifying that a single copy of pJTI/Zeo was integrated to the chromosome 13 locus by Southern blot analysis (Fig. 2A), we selected 2 clones (YX15 and YY9d), each with a single copy in the chromosome 13 hot spot in either the plus or the minus orientation for further evaluation. We found that they maintained the parental BG01V karyotype (data not shown) as well as hESC properties such as typical hESC morphology (Fig. 2B), pluripotency markers Oct4 (Fig. 2C, D), SSEA4 (Fig. 2E), Tra1-60 (Fig. 2F), and Tra1-81 (Fig. 2G), and showed the ability to differentiate into all 3 germ layers (Ref. [23], data not shown).
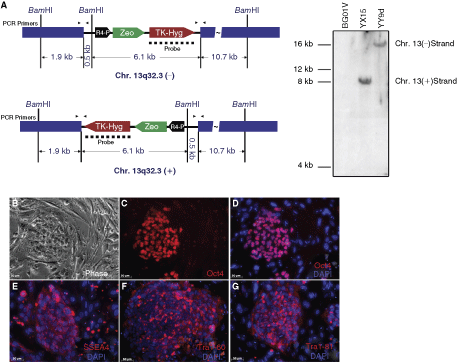
R4 platform lines were correctly targeted to chromosome 13 and maintained pluripotency and self-renewal capacity. (
Retargeting platform lines
A simple targeting vector containing a constitutive EF1α-EmGFP expression cassette was used to retarget the chromosome 13q32 platform site with the R4 integrase. Successful retargeting would result in expression of Zeocin-resistance gene, and therefore allow efficient selection of the desired Zeocin-resistant clones. We obtained an average of 17 colonies per 106 transfected cells (the number of colonies varied from 8 to 30 in multiple experiments). Since all of the surviving retargeted clones should be targeted to the exact same locus, all the clones picked should be identical. As described in Figure 3A–D, all clones tested were targeted to the correct predetermined locus. One clone, however, showed 2 probe-reactive bands in the Southern analysis (Fig. 3D, clone F2BG10) indicating that it either was a single clone containing 2 copies (1 in the target site, and 1 randomly integrated) or resulted from a mixed clone colony (not a single cell clone). In addition to these 7 clones tested, 15 others analyzed by Southern blot were found to have a single copy of the retargeting vector (data not shown). Of the 22 clones that have been analyzed, all had integration events at the predetermined target site and 1 was potentially a double integrant.
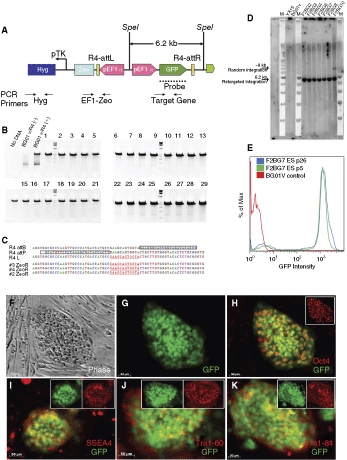
R4 platform lines were successfully retargeted to the predetermined chromosome 13 site using the EF1α-GFP vector. The schematic (
Locus-specific expression in pluripotent cells and after differentiation to multiple lineages
All retargeted clones maintained typical hESC morphology and expression of pluripotency markers (Fig. 3F–K). In addition, after 2.5 months (21 passages) of continuous culture, >95% of these engineered cells continued to express GFP. When quantified with flow cytometric analysis, a mean GFP intensity peaked at ∼103, which was similar to the GFP expression profile for passage 5 (Fig. 3E). This result suggests that the integration is stable through multiple passages and that the chromosome 13 locus is permissive for high expression of transgenes.
We then checked GFP expression of the EF1α-GFP clones upon differentiation to multiple lineages (ectoderm, mesoderm, and endoderm) by randomly differentiating them using an established embryoid body (EB) formation protocol. After 21 days of differentiation, retargeted clones maintained GFP expression as a relatively homogeneous population (Fig. 4). Of all five clones tested, 90%–97% of the undifferentiated cells were GFP+. After 21 days of differentiation (experiments were done in triplicate or quadruplicate), 80%–91% of cells were GFP+. Figure 4A and 4B shows the result from 1 of the 4 repeated experiments for clone F2BG7 (derived from platform line BG01V YX15, chromosome 13 plus strand). Similar results were obtained for retargeted clones using the BG01V YY9d platform line (chromosome13q32 minus strand).
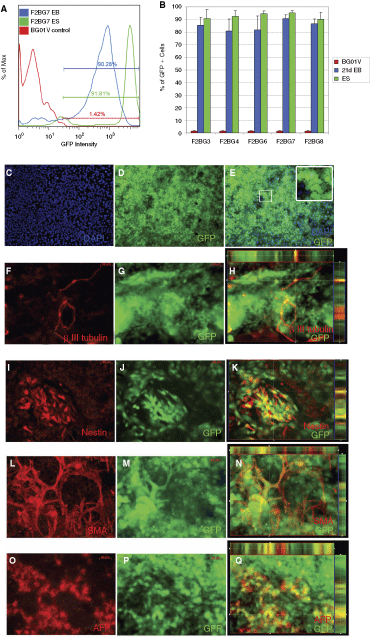
Lack of silencing in EF1α-GFP-retargeted lines after being differentiated into multiple lineages using a 21-day embryoid body protocol. Clone F2BG7, an EF1α-GFP retargeted clone to platform line YX15, showed no appreciable silencing in terms of GFP expression in flow cytometric analyses (
Immunocytochemistry data were consistent with the FACS result. Upon differentiation the majority of cells continued to express GFP (Fig. 4C–E). Cells differentiated toward the ectodermal lineage, as detected by β-III tubulin and Nestin antibody staining, coexpressed GFP as shown by the merged image and Z-stack split views along the
The human Oct4 promoter targeted to the chromosome 13 locus is appropriately regulated
In order to examine whether the chromosome 13 locus allows appropriate transcriptional regulation of heterologous promoters, we integrated an Oct4-GFP cassette into the YX15 platform line. The hypothesis was that if this locus permitted appropriate regulation of the Oct4 promoter as reported by the expression of a transgene (GFP), the engineered line should display GFP expression in the undifferentiated state. Upon differentiation, GFP expression should be down-regulated along with endogenous Oct4 expression. A representative retargeted Oct4-GFP clone showed GFP coexpression with Oct4 in the undifferentiated state (Fig. 5A–D). When this clone was differentiated toward ectodermal, mesodermal, and endodermal lineages, as shown by βIII tubulin (Fig. 5E), Nestin (Fig. 5F), SMA (Fig. 5G), and AFP (Fig. 5H) expression, the GFP expression was appropriately shut down along with the down-regulation of Oct4 expression. Flow cytometric analysis supported this conclusion (Fig. 5I).
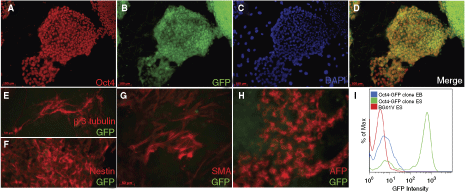
The human Oct4 promoter targeted to the chromosome 13 locus is appropriately regulated. The platform line YX15 containing the R4 system targeted to chromosome 13 was retargeted with an Oct4-GFP vector and measured for GFP expression (
A GFP-expressing population of neural lineage cells could be obtained by directed differentiation of retargeted EF1α-GFP clones
EF1α-GFP-retargeted cells subjected to a directed neural differentiation procedure formed typical neural rosette structures and maintained high expression levels of the GFP reporter (Fig. 6A–C). As expected, these cells lost Oct4 expression (Fig. 6D–F) and uniformly expressed neural stem cell markers Sox1 (Fig. 6G–I), Nestin (Fig. 6J–L), and Musashi1 (Fig. 6M–O). Upon further differentiation, this population gave rise to cells of neuronal lineages, such as βIII tubulin (Fig. 6P) and NF160-expressing neurons, A2B5 immunoreactive glial precursors (Fig. 6Q), and PDGFRα+ oligodendrocyte precursors (Fig. 6R), while continued to express GFP along the different neural differentiation paths. Taken together, these results show that EF1α-GFP clones obtained from the R4 platform lines could be induced into a relatively pure population of GFP-labeled neural stem/precursor cells that could be further differentiated in vitro along the neural lineage pathway.
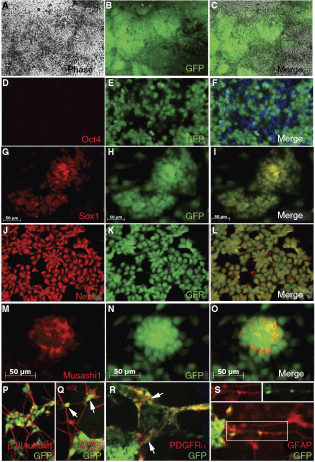
GFP-expressing population of neural lineage cells could be obtained by direct differentiation of retargeted EF1α-GFP clones. A directed neural differentiation protocol was performed on retargeted EF1α-GFP clones and populations of GFP-labeled pure neural progenitor cells were obtained and characterized. After neural differentiation, the cells continued to express GFP (
Generation of platform lines in normal hESC cells
To demonstrate that the retargeting platform we developed would also be useful in karyotypically normal hESC, we extended the technology to H9, a widely used hESC line with normal karyotype. H9 cells were transfected with the plasmid pJTI/Zeo and pCMV-phiC31Int. Since we had already demonstrated that the pseudosite at chromosome 13q32.3 had favorable expression characteristics, H9-derived colonies resistant to hygromycin B were screened by PCR for the presence of the R4 acceptor site at this locus. We successfully targeted the R4 acceptor to the chromosome 13q32.3 locus in five out of 45 clones (11%, Fig. 7A). Of these, 4 were found to have a single copy of the R4 acceptor site (Fig. 7B). All 4 of the single copy lines still maintained expression of hESC markers (Fig. 7D–F) and a normal karyotype (data not shown). One of these clones was retargeted with the plasmid pER4B-EG, and other clones were retargeted to the predetermined chromosome 13 site (Fig. 7C), and expressed GFP in a constitutive manner (Fig. 7G–I).
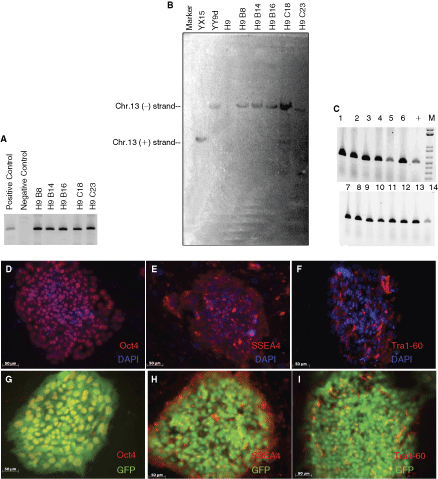
Generation of targeting platform and retargeting line in normal human embryonic stem cells (hESCs) H9. The locations of the PCR primers, restriction sites, and DNA probes are shown in the schematic in Figures 2A and 3A. Five clones derived from H9 were targeted to the hot spot on the minus strand of chromosome 13q32 as examined by PCR screening (
Discussion
Described here is the development of a genomic retargeting system where 2 integrases, phiC31 and R4, were used sequentially to insert expression cassettes into a predetermined locus in the hESC genome. Based on previous mapping information of phiC31 integration sites in hESCs [23] and preliminary retargeting data, we chose 2 BG01V-derived R4 platform lines that were mapped to a single chromosomal locus (13q32, plus and minus strand, respectively) and performed a second round of targeting to insert a GFP expression cassette driven by a constitutive promoter EF1α or a lineage-specific promoter Oct4. We have also demonstrated that platforms targeted to this hot spot can be obtained from the H9 line, which is karyotypically normal.
Since only those cells that bear constructs correctly inserted into the platform site will express the antibiotic resistance gene, clones that survive selection after a second round of targeting should all be identical, making it possible to propagate a pool of clones rather than having to screen them individually for favorable characteristics. Our results were consistent with this hypothesis, with only one of the 22 YX15 platform line (chromosome 13 plus strand)-derived clones containing an additional random integrant (Fig. 3D). This is a seemingly rare event, the overall frequency of which should be vetted out as more clones are created in various target lines.
The karyotype of the majority of clones we isolated after targeting and retargeting was consistent with their parental lines, that is, BG01V and H9. Very occasionally in BG01V, we found retargeted clones that had partial abnormality, and we have not detected any karyotype changes in engineered normal hESC line H9 (Fig. 7). Karyotype drift is a recurring issue in hESC culture, especially after cells are subjected to the stress of transfection or single cell cloning [32]. However, it is possible that these karyotype abnormalities could have been caused by expression of phiC31 and R4 integrases in these cells [33]. A recent study reported that transformed cell lines containing stably integrated phiC31 integrase expression cassettes were found to contain chromosomal aberrations that might be attributed to promiscuous activity of constitutively expressed integrase [34]. With this in mind, we examined the genomes of both of platform lines described here and their corresponding retargeted clones by PCR and found no evidence of the integration the plasmids expressing either phiC31 or R4 integrase (see Materials and Methods, data not shown). In addition, our long-term culture data confirmed that the targeted and retargeted clones maintained a parental karyotype and stable expression of transgenes. Therefore, although we cannot exclude the possibility of transient integrase expression causing the rare and partial nonparental karyotype results we have found in a few of the engineered clones, we do not believe it is of major consequence. These events were rare, and did not arise at a frequency higher than that obtained during normal culture of hESC lines. We were able to obtain platform lines and retargeted clones with the parental karyotype in every instance, without the need to screen a large number of clones.
In this report, we have focused on the targeting and validation of 2 simple reporters, which were assembled using a 2-fragment Multisite Gateway process. Our laboratory and others [16,18,35], however, routinely assemble multigene constructs up to 8 fragments and as large as 13 kb using this relatively straightforward cloning process. In recent preliminary experiments, we have successfully inserted a 13-kb 2-color reporter cassette consisting of 2 promoters driving reporter genes GFP and tagRFP into the chromosome 13 locus. The effective payload (expression elements) consisted of ∼10 kb, which represents an improvement over what is currently possible using lentiviral and other viral approaches where the payload limit is as low as 6–8 kb. These constructs were targeted with similar efficiency as the smaller EF1α-GFP constructs (data not shown), suggesting that the upper size limit of this system has not been reached.
We have shown that the chromosome 13 locus remains transcriptionally active upon differentiation into any of the 3 germ layers during random EB formation. Further, we were able to obtain an essentially pure population of GFP-labeled neural stem/precursor cells and mature neural cells using the EF1α-GFP clone targeted to the chromosome 13 locus (Fig. 6). Our previous results suggest that several additional potential sites are available, including sites on chromosomes 2, 6, 10, 17, and 21 [23]. The properties of these loci are currently under investigation. Recently, several reports have documented other possible sites in the hESC genome that are potentially useful for this sort of engineering, including the “Envy” locus on chromosome 12 [36] and the human Rosa 26 locus [37] on chromosome 3. In the latter study, Irion et al. used homologous recombination to generate a target/reporter hESC line by “trapping” the ROSA26 promoter and placing a receptor site at that locus. They then used a recombinase-mediated cassette exchange (RMCE)-based method to target a genetic element to that site [37]. The integrase-mediated targeting strategy described in the present work complements the Irion et al. study in that we have targeted and identified several other potentially useful genomic loci in the hESCs and have shown that at least one of them (chromosome 13q32) can be retargeted with high efficiency. Furthermore, we show a significant advantage in targeting efficiency with the R4 integrase-mediated system since essentially 100% of the drug-resistant colonies obtained from the targeting reaction result from a true targeting event, therefore selection of properly targeted clones does not require cell sorting. These 2 systems are not mutually exclusive, however, as one may anticipate configurations where combinations of locus selection and locus retargeting technologies could be used. Further, evaluation of these and other loci and technologies may add considerable versatility to future versions of this and similar hESC engineering platforms. By using the technology described here, either alone or in combination with other methods, one can anticipate the creation of a hESC platform line containing multiple platform sites at separate loci. This would increase the amount of genetic payload that could be specifically delivered to the cells and enable higher order complexity of assays in a single cell line.
Successful development of efficient targeted integration systems like the one described earlier could facilitate the generation of platforms such as single or multiple lineage reporter cell lines for in vitro differentiation studies and transplant tracking in vivo as well as cell-based drug screening assays in stem and progenitor cells and terminally differentiated progeny. The latter would likely be more consistent and relevant than assay systems currently used in human and rodent primary and transformed cell models. In addition to specific reporter functions, this system will be useful for controllable, stoichiometric expression of cell perturbation molecules including shRNA, miRNA, dominant negative signaling proteins. Since each cell in a population would carry a single copy of the perturbation construct at identical loci, the dosage of perturbation agent would be expected to be homogeneous leading to a similar resultant phenotype from cell to cell. These tools can also be afforded more versatility by combining with them existing useful technologies such as Cre/lox or Flp/frt integration and excision for in situ modulation of the assay system. Finally, given that cells can be targeted to a specific locus, matched allelic series of related assay lines can be created and these can be compared more reliably since each assay construct would be expressed in the same cellular background (same copy number and locus in all cells).
In the future, systems similar to this could be useful as platforms for creation of engineered cell lines as therapeutic tools in extracorporeal devices and for cell or tissue replacement. Although any genetic manipulation will make the regulatory burden higher than with using unmodified cells, the R4-retargeting technology described here, or something similar, may facilitate the use of engineered cells in the clinic by allowing the targeting of a specific, validated site in the genome rather than randomly integrating in a potentially damaging way. It has already been demonstrated that phiC31 could have tremendous potential in gene therapy applications [38].
In summary, we have created a platform that facilitates genetic engineering of hESCs in generating reporter lines, signaling pathway assay systems, and creating disease models, therefore providing potential sources for basic research on developmental biology, in drug screening, and perhaps eventually cell-based therapy.
Footnotes
Acknowledgment
This work was supported by Invitrogen Corporation, part of Life Technologies Corporation.
Author Disclosure Statement
All authors declare that they have no potential conflict of interest in connection with the submitted article.