
Abstract
Aging is an unavoidable process associated with a progressive decline of muscle mass, strength, and regenerative ability. Satellite cells are a muscle stem cell (MuSC) population that plays a key role in mammalian muscle regeneration, by awakening from quiescence and then migrating to sites of damage, expanding in number to generate progenitor cells, and then either differentiating to rebuild the muscle tissue or self-renewing to repopulate the stem cell pool. Emerging evidence suggests that the aging process impairs the activation potential and the regenerative capacity of MuSCs. This review explores some of the recent discoveries of how mis-regulation of intrinsic and extrinsic mechanisms drive the decline of MuSC function in aging muscles, and we discuss new strategies to rejuvenate aged MuSC function for regenerative medicine. Understanding these processes will speed up the development of novel therapeutics for counteracting muscle loss and improve muscle healing in the elderly.
Introduction
The significant increase in life expectancy over the past 75 years is one of humanity’s most extraordinary milestones, rising by 58%, from 46.4 to 73.5 years. 1 However, with longer lives comes the challenge of maintaining good health, particularly mobility, which represents a key factor in preserving independence and daily function in aging individuals. 2 As we get older, our skeletal muscle gets weaker, smaller, and gradually loses its ability to heal efficiently after injury, which increases the risk of falls and fractures, ultimately, leading to severe disability or even death. 3 Therefore, it is essential to develop new therapeutics to counteract the negative effects of aging on the skeletal muscle system.
In this review, we discuss the loss of muscle healing in aged vertebrate animals with a focus on how deficiencies in muscle stem cell (MuSC) activity contribute to the muscle dysfunction associated with aging. Additionally, we provide an overview of both pharmacological and nonpharmacological interventions to prevent or combat muscle aging in the elderly.
Role of MuSC activation in healthy regeneration and aging
Skeletal muscle is one of the most regenerative tissues in the mammalian body due to a MuSC population known as satellite cells. 4 In 1961, a population of cells situated between the basal lamina and the plasma membrane of muscle fibers was hypothesized to be essential for muscle regeneration, 5 a theory firmly established later on when mice lacking the paired box 7 protein (Pax7)—a critical determinant of the MuSC fate 6 —in MuSCs were shown to have a complete failure of muscle repair. 5,7 –9
In the years following, several decades of work would show that MuSCs transition through distinct cellular states known as quiescence, activation, differentiation, and self-renewal to maintain muscle integrity and support regeneration. 10,11 Quiescent MuSCs express PAX7 and initially exhibit a nondividing state before tissue damage. 12 In response to muscle injury, MuSCs induce the expression of myogenic determination 1 (MYOD1) and myogenic regulatory factor 5 (MYF5), and then over the next 2–3 days exit quiescence and initiate the first cell division (i.e., the rate-limiting step 13 ). Subsequently, MuSCs will either continue proliferating as myoblasts or start to differentiate by expressing Myogenin (MYOG) followed by fusion with each other to form de novo fibers or with existing myofibers to repair the damage. 14 Importantly, a select group of proliferating myoblasts “hits the reset button” by upregulating PAX7 while downregulating MYF5 and MYOD1, and undergoing self-renewal back to the quiescent state to replenish the stem cell pool. 6 The delicate balance between myogenic commitment and stem cell preservation ensures that the skeletal muscle tissue remains resilient, adapts to stress, and maintains muscle strength over time. 15
MuSCs are not the only cell types and niche signals in the muscle microenvironment that are needed for efficient muscle regeneration. 16 Indeed, in addition to MuSCs, interstitial cells including endothelial cells, fibro-adipogenic progenitors (FAPs) and a variety of immune cells work together to facilitate muscle repair. 16 Recent data have shown that various cells in the muscle niche secrete signaling factors that guide MuSC activation and the regenerative process. 17 Beyond the local microenvironment, the systemic niche, which comprises circulating hormones and other metabolic factors, also influences MuSC function. 18 The interplay between the local and systemic niches ensures that MuSC activation, differentiation, and self-renewal are finely regulated in response to both tissue-specific needs and physiological cues.
In recent years, it has become clear that aged MuSCs lose the ability to activate properly leading to diminished regenerative ability and thus contributing to the muscle dysfunction associated with aging. 21 –25 In this review, we will discuss some of the most recent mechanisms responsible for the reduced function of aged MuSCs, with an emphasis on mis-regulation of intrinsic (i.e., cell-autonomous) as well as external signals including local niche and blood-borne systemic factors. We will also discuss some of the therapeutic avenues that may be leveraged to combat age-associated skeletal muscle decline. 19
Intrinsic Mechanisms Driving MuSC Dysfunction
Transcriptional and epigenetic alterations
In recent years, advancements in genomic technologies and their ability to capture rare primary cell populations have transformed our views on how adult MuSCs activate and the gene regulatory changes that occur with age that hinder their functional ability. Several recent studies have demonstrated that as MuSCs age, they tend to lose global heterochromatin (with decreases in H3K9me2 and recruitment of Hp1a) and, as a consequence, display more open chromatin regions across the genome, 20 –22 suggesting that a failure to properly control gene expression programs drives the aging MuSC phenotype. What is the cause of these changes? Interestingly, a recent study revealed that MuSCs exhibit reduced levels of S-adenosyl methionine (SAM), which is our cells’ major methyl donor for DNA-specific methylation. It would be expected that lower levels of SAM likely contribute to the loss of silenced DNA (i.e., methylation) that is characteristic of heterochromatin. Indeed, administering SAM to aged and control mice led to the re-establishment of heterochromatin in MuSCs and a boosted muscle repair response in aged mice, 22 demonstrating how environmental metabolites can impact critical epigenetic processes in the nucleus of aging MuSCs. In addition to environmental metabolites such as SAM, the epigenetic state of aged MuSCs can also be reversed by exposure to a youthful microenvironment. Notably, transplantation of aged MuSCs into young recipient mice led to changes in chromatin accessibility and DNA methylation in a subset of genes to a more youthful state. 23 This finding underscores the critical role of the niche in modulating MuSC function—a concept that will be further explored in the section “Dysfunction of local niche signals in aging muscles.”
Considering that transcription factors (TFs) are known to bind accessible regions (including promoters and enhancers) and that aged MuSCs display more open chromatin regions when compared with young MuSCs, 20 it is not surprising that several TFs have recently been shown to contribute to the misfiring of deleterious gene expression programs that promote stem cell dysfunction in aged MuSCs. In one study, global histone proteomics revealed a shift toward more H3 and H4 acetylation in aged MuSCs (signifying more permission chromatin), which led to overexpression of HOX9A, a developmental TF that was shown to turn on known MuSC inhibitors including Wnt, 24 transforming growth factor beta (TGF-β), 25 Janus kinase/signal transducer and activator of transcription (JAK/STAT), 26,27 and other regulators of senescence. 28 In addition to HOX9A, another study found that SLUG, a zinc-finger TF, normally represses p16ink4a (a senescence master regulator) in adult MuSCs, but in aging MuSCs, it is downregulated, 29 providing a more direct link between transcriptional misfiring with a canonical aging phenotype, senescence.
The Notch signaling pathway has been known for decades to be suppressed in aged MuSCs, and restoring its activity in aged MuSCs improves their function and repair ability. 30,31 More recently, a follow-up study revealed that aged MuSCs undergo a failure in mitosis, known as mitotic catastrophe, in part due to loss of Notch-p53 signaling (including the RBPJ TF), 32 likely explaining why an increasing number of MuSCs become less resilient in older muscle. Another study found that the TF FoxO is upregulated in quiescent MuSCs from young muscle tissue, where it mediates both the transcriptional activation of genes involved in maintaining quiescence (CD34 and Igfbp4) and the repression of critical genes promoting cell differentiation (i.e., MYOG). This finding aligns with FoxO depletion experiments in FoxO1,3a,4ΔPax7ER mice, in which MuSCs displayed spontaneous activation and differentiation. Interestingly, MuSCs isolated from geriatric mice (>28 months old) show downregulation of FoxO, suggesting that their seemingly enhanced activation (i.e., initial opening of chromatin) may be dependent on FoxO mis-regulation. 33 A recent study uncovered an additional role for FoxO in promoting autophagy through the inhibition of Deaf1, a repressor of autophagy, thereby facilitating the transcriptional activation of autophagy-related genes. Notably, aged muscle in both flies and mice displays elevated levels of Deaf1. Consistent with this, the autophagy impairment observed in aged muscle, characterized by the accumulation of damaged mitochondria, increased reactive oxygen species (ROS), DNA damage, cellular senescence, and apoptosis, can be attributed in part to the age-associated downregulation of FoxO. 34,35
Adding to this emerging picture, levels of the RNA splicing factor, U1 small nuclear RNA (U1 snRNA), is elevated in aged MuSCs, which promotes splicing to include the long CD47 isoform and resulting in its surface expression, while the short isoform, prevalent in young MuSCs, is retained in the endoplasmic reticulum. 36 The shift from CD47short to CD47long contributes to MuSC dysfunction, since CD47high cells display increased differentiation but reduced self-renewal, while CD47low cells have robust proliferative and self-renewal potential. Indeed, targeting U1 snRNA with antisense morpholino oligonucleotides reduces expression of the long CD47 isoform and improves engraftment of aged MuSCs after transplantation. Mechanistically, CD47high MuSCs secrete high levels of thrombospondin-1 (THBS1), a CD47 ligand that, in aged muscle, inhibits expansion of the CD47low population. Furthermore, blocking THBS1 with a neutralizing antibody increased MuSC numbers (at 0 and 3 days postinjury), myofiber size, grip strength (10 days postinjury), and embryonic myosin heavy chain (eMyHC) expression (6 days postinjury). 36
Moving forward, it will be interesting to see how future dietary supplements, drugs, and/or exercises can restore heterochromatin, transcriptional programs, and RNA processing pathways to re-establish balance in the epigenetic and transcriptional circuits, and thus, programming more youthful states in older MuSCs.
Senescence
Senescence is a cellular state characterized by cell cycle arrest, DNA damage, chromatin remodeling, and upregulation of proinflammatory cytokines known as the senescence-associated secretory pathway (SASP). 37 In senescent cells, growth arrest is typically driven by the activation of two main tumor suppressor pathways: p16INK4a/Rb and p53/p21CIP1. 38,39 Previously, it was found that MuSCs from extremely old mice (>28 months old) undergo senescence that is triggered by the increased expression of the tumor suppressor gene, p16Ink4a. As a consequence, geriatric MuSCs fail to activate, expand the progenitor cell population, and regenerate muscles after chemical injury in vivo, and repressing p16Ink4a in geriatric MuSCs is sufficient to restore proper repair activity. 39
Building on these findings, to uncover the repertoire of muscle-resident senescent cells in vivo before and after myo-trauma in young and aged mice, a group took advantage of a new p16-3MR mouse model, in which they expressed monomeric red fluorescent protein (RFP) along with thymidine kinase under the p16 promoter. 40 Remarkably, they found that while senescent cells (RFP+ve) were low in uninjured muscles, within 3 days after cardiotoxin (CTX) injury, they increased after 3 days but then decreased by 7 days, suggesting a transient senescent state even in young adult mice. Interestingly, old muscles displayed a longer period of time, whereas RFP+ve senescent cells persisted in the muscle after injury. 40 Furthermore, through scRNA-seq, they found many RFP+ve senescent cells among MuSCs, FAPs, and blood cells and that RFP+ve cells expressed cytokines (i.e., SASP) that when secreted into the local microenvironment created an “inflammaging” signature that impedes muscle regeneration. 40 From a functional standpoint, it was found that removing senescent cells in young and old mice via administration of Ganciclovir (i.e., eliminating RFP+ve cells) to p16-3MR mice, administration of two commonly used senolytics called Dasatinib and Quercetin validated by a previous study, 41 or reducing the secretome of senescent cells using a CD36 neutralizing antibody was sufficient to restore proper stem cell function and muscle healing to aged mice. 40
Metabolic dysfunction
Given how critical metabolic activities are in normal cellular behavior, 42 it is not surprising that metabolic malfunction plays a crucial role in the declining activity of aging MuSCs. 43 –45 Typically, quiescent MuSCs are thought to have low energetic demands, reduced transcription of RNAs, and lower protein synthesis. 46,47 In adult MuSCs, fatty acid oxidation is thought to be the main metabolic pathway that MuSCs rely on during quiescence. However, as they transition to the activated state, their metabolism switches toward glycolysis, while oxidative phosphorylation (OXPHOS) becomes more predominant during differentiation. 44,48,49 Interestingly, aged MuSCs display a switch from OXPHOS to glycolysis, 45 concomitant with their reduction in differentiation kinetics. 50 Thus, it appears that fully activated and differentiating MuSCs require glycolysis and OXPHOS to meet the energetic demands for breaking quiescence, respectively.
Consistently, a recent study found that adult MuSCs displayed an increase in mitochondrial fission genes, including Drp1 and Mff, and had smaller, less complex mitochondria 3 days after in vivo injury, suggesting a role in mitochondrial fission (i.e., split of one mitochondria into two) in facilitating MuSC activation. 51 When the mitochondrial fission protein Drp1 was genetically removed from MuSCs, (1) mis-formed mitochondria (e.g., having abnormal cristae) were found in FACS-isolated adult MuSCs visualized by electron microscopy; (2) fewer YFP+ve (Ki67+ve) proliferating myogenic cells were found; and (3) smaller eMyHC-positive fibers were observed after 7- and 14-days postinjury. What is the link between mitochondrial fission, OXPHOS, and aging MuSCs? Apparently, without Drp1 in MuSCs, these cells fail to turn on critical electron transport and TCA cycle genes needed for OXPHOS. Finally, they found that aged MuSCs have reduced Drp1, smaller mitochondria, and when Drp1 was overexpressed in aged MuSCs via lentiviral delivery, it restored their proliferative and in vivo engraftment ability. 51 Another key protein involved in maintaining mitochondrial health and function that is downregulated in aged MuSCs relative to young MuSCs is α-Klotho. 52 siRNA-mediated knockdown experiments revealed that α-Klotho is essential for maintaining OXPHOS. Its loss leads to reduced OXPHOS activity, disruption of mitochondrial cristae, and increased mitochondrial DNA (mtDNA) damage. Notably, α-Klotho supplementation restored mtDNA integrity and mitochondrial bioenergetics in aged MuSCs to youthful levels in vitro. On the contrary, in vivo, it enhanced functional muscle regeneration, as seen by an increase in centrally nucleated regenerating fibers and myofiber cross-sectional area (CSA). 52
In addition to mitochondrial factors and signaling pathways that mediate mitochondrial biology, enzymes that impact how mitochondria deal with oxidative stress have also emerged as a critical determinant of aging. For example, it was recently found that the majority of young MuSCs exhibit a high glutathione metabolism, which has a robust proliferative and regenerative response. However, with aging, this once single population splits into two: a reduced glutathione (GSH)low and a GSHhigh population. GSHlow MuSCs exhibit impaired activation, reduced mitochondrial respiration, and elevated ROS due to increased Nuclear Factor kappa B (NF-κB) and Smad3 signaling. Interestingly, NF-κB inhibition or N-acetylcysteine (a GSH precursor) treatment can convert GSHlow into GSHhigh cells in aging animals. These findings highlight redox modulation as a promising therapeutic strategy to restore MuSC function in aging muscle. 53
One of the most critical vitamins in skeletal muscle health is nicotinamide adenine dinucleotide (NAD+), and its function in MuSCs has recently taken center stage in aging research. 54 NAD+ is a cofactor for a family of mono-ADP-ribosyl-transferases (MARylators), poly-ADP-ribosyl-transferases (PARylators), and Sirtuins. NAD is metabolized by these enzymes, resulting in the addition of either a single ADP ribose or a chain of ADP ribose to target protein substrates. 55 It was shown that NAD+ levels are elevated in early activated MuSCs but then decline as they transition out to the fully activated, dividing state (3 days in culture). 44 Collectively, two groups then showed that a key NAD+-consuming enzyme, Sirtuin 1 (Sirt1), promotes autophagy, which appears to provide the energy to enable MuSCs to grow in size and metabolic activity necessary for proper MuSC activation and regeneration. 44,56 Another study found that the transcriptional regulator FOS activates the ecto-enzyme Adp-Ribosyl-transferase 1 (Art1) and disruption of Art1 via shRNAs or small-molecule inhibition of its MARylation activity was sufficient to reduce the activation potential of adult MuSCs. 57 However, despite these few examples, how ADP-ribosylation family members impact activation of MuSCs and regeneration in vivo is largely unexplored. Nonetheless, what is absolutely clear is that NAD+ levels decline substantially with age, and boosting NAD+ levels systemically by administering either NAD (NMN) or nicotinamide riboside (NR) has been reported to enhance MuSC activity and muscle health in aged mice. 43 Future work to identify the critical NAD+-consuming enzymes that facilitate the beneficial effects of NAD-supplementation on muscle healing is needed to translate these findings successfully in the clinic.
Dysfunction of Local Niche Signals in Aging Muscles
Fibro-adipogenic progenitors
FAPs are a heterogeneous mesenchymal stem cell population that resides in the interstitial space of skeletal muscle and are capable of generating both fibroblast and adipocyte populations. 58,59 Two back-to-back studies showed that the number of FAPs increases rapidly after muscle injury in vivo, while the other showed that coculture of FAPs with myoblasts increased myoblast differentiation. 60,61 Direct evidence for FAPs role in MuSC function came when a group generated a PDGFRαCreER mouse and crossed it to a diphtheria toxin (Dtr) mouse, such that all cells expressing PDGFRα would be eliminated in vivo. Using this system, they showed that removing PDGFRα cells in vivo led to an impairment in MuSC expansion, regeneration, and force generation after myo-injury, providing the first direct evidence of their essential functions in coordinating MuSC activation and muscle repair in vivo. 62
One of the major mechanisms proposed to explain how FAPs impact MuSC function during adult regeneration is by secreting signaling molecules into the local microenvironment that stimulate MuSC function. A recent study showed that WISP1, a signaling protein that enhances MuSC function in adults, is downregulated in aged MuSCs. 63 Also, they found that adding back WISP1 is sufficient to recover the loss of regenerative activity of aged MuSCs. 63 In addition to influencing MuSC behaviors directly, another study found that FAPs are a major source of interleukin-33 (IL-33), which is necessary for the efficient recruitment of regulatory T cells (Tregs). Interestingly, since activated FAPs have also been shown to decline in number with age, less FAPs results in less IL-33 secreted into the injured microenvironment, ultimately leading to diminished muscle regeneration. 64
Immune cells
Skeletal muscle regeneration after injury is significantly impacted by immune cell infiltration, including neutrophils, macrophages, and T lymphocytes. 65 Neutrophils are the first responders, entering the injured muscle and releasing cytokines such as interferon-gamma (IFN-γ) and TNF-α, which in turn recruit macrophages. 66,67 Coculturing macrophages with MuSCs ex vivo led to increased expression of MyoD, suggesting that macrophages promote MuSC activation in addition to their more traditional role in clearing tissue debris through phagocytosis. 68 Consistent with these ex vivo studies, when macrophages were depleted in vivo using injections of an antibody targeting the macrophage-colony stimulating factor (i.e., resulting in the inhibition of macrophage proliferation as reported previously 69 ), this led to reduced MuSC proliferation, fewer eMyHC+ve fibers after injury, and increased fibrosis in the muscle tissue. 70 Following macrophage infiltration into the damaged tissue, T lymphocytes migrate into the injury site between 3- and 5-days postinjury. Ex vivo studies demonstrate that T lymphocytes can enhance the expansion of murine MuSCs by secreting proinflammatory cytokines, such as IL-1α, IL-13, TNF-α, and IFN-γ, and promoting the migration of MuSCs isolated from young rats. 71,72 Lastly, Tregs, a subset of T lymphocytes, begin to accumulate in the damaged muscle around 4 days after injury and remain for up to a month. 73 Tregs participate in the switch of macrophages from M1 proinflammatory to M2 anti-inflammatory and stimulate MuSCs proliferation and migration mainly through the release of amphiregulin. 73,74 Cytokine production by Tregs, especially IFN-γ, IL-6, and TNF-α, has been found to regulate MuSC activation, proliferation, and differentiation. 65
Studies on aging muscle regeneration have revealed a strong link between impaired muscle repair and reduced recruitment of immune cells, particularly macrophages and T lymphocytes, as any disruption in leukocyte recruitment from the bloodstream to the muscle limits their contribution to muscle regeneration. 64,75,76 A recent study provided a detailed analysis of the immune cell types in young and aged muscle tissues during injury, revealing a significant heterogeneity within the resident macrophage population. 77 This work and others identified an enrichment of “proinflammatory” Ly6Chigh macrophages and a reduction in Ly6Clow “repair” macrophages in the aged muscle tissue. 76 –78 This imbalance leads to lower levels of cytokines and growth factors, including growth differentiation factor 3 (GDF3) and mesencephalic astrocyte-derived neurotrophic factor (MANF) secreted by repair macrophages. Interestingly, restoration of GDF3 and MANF levels in aged muscles has been shown to improve regeneration. 76,78 Notably, the proportion of Ly6Clow macrophages in aged mice can be restored to youthful levels by administration of Meteorin-like (Metrnl), a peptide secreted by macrophages during the early phases of muscle injury. Metrnl expression declines with age, but its supplementation in aged mice has been shown to improve muscle regeneration, as shown by increased CSA of myofibers. Furthermore, Metrnl facilitates the clearance of FAPs by stimulating macrophage-derived TNF-α production, which induces FAP apoptosis and limits their age-associated fibrogenic conversion. 79 Another recent study using scRNA-seq of 3-day CTX-injured young muscle identified a novel population of IFN-responsive macrophages, termed IFNRMs, which were significantly reduced in injured muscles of aged mice. 80 IFNRMs were the main producers of CXCL10, a cytokine that promotes MuSC proliferation (measured by EdU incorporation) and differentiation (measured by fusion index) in vitro. Interestingly, injecting CXCL10 into the injured muscles of aged mice was able to boost the number of proliferating MuSCs and improve muscle fiber regeneration. 81 Another scRNA-seq analysis on murine hindlimb muscles revealed that aged muscles have reductions in mRNA for proteins involved in chemotaxis of granulocytes and monocytes (Cxcl1, Cxcl2, Ccl2, Ccl7), response to IFN-γ (Cxcl10, TNF, Zfp36), and anti-inflammatory M2 macrophage markers (Lyve1, Folr2, Mrc). On the contrary, genes related to cellular detoxification (Gsr, Hp, Prdx1, Prdx5, Prdx6), inflammation (Fabp4, Il1b), and senescence (Gpnmb, Spp1) were upregulated, suggesting that mis-regulation of the immune cell milieu is a general feature of aged muscles. 80
Lastly, a decline in Treg cell numbers in aged muscles is driven by defects in their recruitment, reduced proliferation, and impaired retention within the injured tissue. 64 A rescue of Treg cell proliferation was observed in 22-month-old mice treated with IL-33 at the time of cryoinjury, which increased CSA of injured muscle fibers in vivo and increased the efficiency of colony formation in vitro. 64 These findings in mouse models have been corroborated by clinical and preclinical studies showing an age-related reduction in T cell populations, particularly CD4+CD28− T cells, which is associated with the onset of sarcopenia—defined as a skeletal muscle mass index lower than 7.26 kg/m2 in males and 5.45 kg/m2 in females (as previously reported in Refs. 82,83 ).
Neuromuscular junctions
Neuromuscular junctions (NMJ) are synaptic connections between the innervating motor neuron and muscle fiber sarcolemma and recent work has revealed that mis-regulation between MuSCs and NMJs contributes to the aging phenotype. 84,85 NMJ are required to initiate the action potentials necessary for muscle contractions and maintaining proper muscle health overall. 84 A sequence of studies revealed that MuSC depletion induced muscle atrophy (smaller fibers), fibrosis, a fiber type switch from IIB to IIA, reduced force generation of skeletal muscle, and a failure to fully regenerate NMJ structure at postsynaptic myonuclei after a sciatic nerve injury—deficits often observed in aged muscles. 86 These data suggest that MuSC and NMJ cross-talk is essential for maintaining proper muscle size and function as we age. A follow-up study showed that as mice age, the NMJs degenerate, which they attribute to a propensity to lose the functional MuSCs population. Interestingly, forcibly expressing Sprouty1 (Spry1) was sufficient to restore the MuSC population and effectively mitigated NMJ deterioration, thereby improving muscle force generation and function. 85,87 –89
Mis-Regulation of Systemic Factors
Blood-borne factors
One of the first pieces of evidence arguing that the systemic environment has a significant impact on driving the aging phenotype came 20+ years ago, when a group transplanted young muscle into old hosts and old muscle in young hosts, ultimately discovering that aged muscles in a young host regenerated efficiently, while young muscles in an aged host regenerated poorly. 90,91 Given that vasculature is a critical component of the niche in skeletal muscle, these data pointed toward the presence of circulating systemic factors that program MuSCs with either youthful or older properties. 31 Based on these assumptions, a series of studies showed that when you connect the circulatory systems of young and aged mice (i.e., heterochronic parabiosis) and examined muscle regeneration in comparison with young–young and old–old parabionts (i.e., the isochronic controls), that exposure to a youthful environment was sufficient to restore stem cell activation (BrdU incorporation and Notch signaling), muscle repair (assayed by eMyHC staining at 5 days), and lessen fibrosis in the aged MuSCs and muscle tissue. 24,31 Interestingly, the beneficial effects of parabiosis appear to extend beyond skeletal muscle by also improving liver and brain function. 31,92 These data indicated that the beneficial effect of heterochronic parabiosis on aged MuSCs is attributed to either (1) a transfer of young blood-borne factors that instruct youthful fates in aged MuSC or (2) that there are pro-aging molecules in old circulation that are diluted when exposed to a youthful environment. These two models were tested directly when a recent study engineered a system to exchange young and old blood directly into young and old mice removing the confounding effects of parabiosis. 93 In series of studies, it was first shown that transfer of young blood into old mice enhanced the muscle regenerative abilities of the older mice 93 similar to what older parabionts experienced when connected to a young mouse in heterochronic parabiosis. 24,31,92 In a key second study, they even replaced young blood with 5% albumin in saline, which completely removes the contribution of young blood, and they still observed a beneficial effect of the blood transfer on aged muscles. 94 While it seems likely that pro-regenerative, youthful factors likely play a role in maintaining muscle health, 95,96 these data suggest that the dilution of old blood, and specific factors like beta-2-microglobulin (B2M) 93 and TFG-β, 94 as having the greatest contribution on the health of aged mouse muscle.
Strategies for Rejuvenating MuSC Function in Aged Skeletal Muscle
Multiple therapeutic strategies are currently being explored to rejuvenate aged MuSCs. These approaches include exercise, TF reprogramming, dietary and pharmacological interventions, which are discussed below. A comparative overview of these strategies is provided in Table 1.
Therapeutic Strategies for MuSC Rejuvenation
AMPK, AMP-activated protein kinase; MuSC, muscle stem cell; NAD+, nicotinamide adenine dinucleotide; OKSM, OCT4, KLF4, SOX2, and MYC; OXPHOS, oxidative phosphorylation; TGF-β, transforming growth factor beta.
Exercise
Exercise is emerging as potentially one of the most effective, nonpharmacological strategies for improving MuSC function in aged skeletal muscles. 97,98 It has been known for decades that exercise can enhance the activation potential, and thus regenerative ability, of MuSCs in response to myo-injury. 99 From an experimental perspective, the two common types of exercise assays in the laboratory are in-house voluntary wheel running or involuntary treadmill running.
Voluntary exercise typically involves moderate, self-regulated activity in which each subject can control the intensity and duration of the exercise. It was recently found that when adult mice performed 3 weeks of nonstrenuous, voluntary wheel running, exercised mice had more eMyHC+ve fibers 7 days after BaCl2 injury when compared with control mice, suggesting enhanced muscle healing. 100 To test whether the benefits of exercise were transferred to MuSCs as an intrinsic mechanism, they transplanted MuSCs from exercised and nonexercised mice and found that MuSCs from exercised mice displayed greater transplantation efficiency at 10 days postinjury in vivo, and when cultured ex vivo, they incorporated more EdU and had higher survival, indicating that the exercised environment has a long-lasting benefit on aged MuSCs. Next, using transcriptomic analysis, they identified Cyclin D1 (Ccnd1) as a key intrinsic target that is activated in MuSCs from old, exercised mice. Finally, using several MuSC-specific overexpression mouse models, they showed that elevating Ccnd1 in old MuSCs restored their activation potential and repair ability after injury in vivo; however, this effect was lost when old MuSCs overexpressed a catalytically dead Ccnd1 (K112E). 100 Altogether, these results show how voluntary exercise appears to be the most accessible way to rejuvenate MuSC function and preserve healing ability in the elderly even if only a transient effect. 100 Consistent with this observation, another study showed that moderate-intensity treadmill running in aged mice has been shown to increase MuSC numbers approximately 1.6-fold over 8 weeks. 101 Beyond activating MuSCs, recent findings suggest that voluntary exercise can also rejuvenate the aged muscle niche through a reduction of age-associated inflammatory pathways, particularly the TNF-α pathway in muscle monocytes and the IFN-γ pathway in T lymphocytes. 98
On the contrary, high-intensity exercise, including heavy resistance training, places significant stress on muscles, which can lead to muscle damage. 102 However, the intensity of training is positively correlated with an increase in MuSC numbers. 103,104 In a study combining endurance and resistance training over 14 weeks involving healthy 73-year-old men, a 38% increase in MuSCs (identified as NCAM+ve via immunohistochemistry) was observed, primarily in type II fast-twitch fibers of the vastus lateralis and deltoid muscles. 103 Interestingly, a decrease in MuSC numbers in the elderly has been proposed as a key factor in the etiology of type II muscle fiber atrophy in aging, a hallmark of sarcopenia, and high-intensity exercise was able to reverse this decline. 104 Moreover, the increase in MuSC numbers following high-intensity exercise contributes to hypertrophy by adding new myonuclei to existing muscle fibers. 105 Therefore, high-intensity exercise is also a crucial strategy for preventing muscle wasting, as it boosts MuSC numbers, stimulates muscle hypertrophy, and increases muscle strength in the elderly. 106
Dietary interventions
Currently, a combined approach of exercise and nutritional supplementation is being explored to improve the function of aging muscles. Evidence suggests that adequate intake of protein, vitamin D, and antioxidants may contribute to improved muscle health in sarcopenic individuals. 107
Vitamin D plays a role in maintaining skeletal muscle health. Vitamin D deficiency is associated with musculoskeletal diseases, including bone-related conditions such as osteoporosis and osteomalacia, as well as muscle-related diseases including sarcopenia. 108 A meta-analysis of 30 clinical trials involving 5165 participants, 72% of whom were women with a mean age of 61, examined the effects of varying doses of vitamin D on muscle strength. The results were mixed, with supplementation below 30 nmol/L showing significant improvements in muscle strength only in individuals aged 65 years or older. 109 Notably, MuSCs, but not the adult skeletal muscle, express the vitamin D receptor, suggesting a role for vitamin D in muscle progenitor cell function. 110 However, the effects of vitamin D on myoblast proliferation and differentiation remain controversial. Several studies have reported inhibitory effects on C2C12 myoblast proliferation. 110 –112 A recent work showed that vitamin D inhibited both myoblast proliferation and differentiation and promoted quiescence through FoxO3 induction and Notch signaling activation in human myoblast cultures exposed to vitamin D for 24 hours. 113 Conversely, another work demonstrated enhanced differentiation in primary murine myoblast cultures treated with active vitamin D, a finding supported by similar results in human myoblasts. 114,115 Therefore, further research is warranted to elucidate the precise mechanisms and beneficial effects of vitamin D on aging MuSCs and more broadly, skeletal muscle.
NAD+ is a coenzyme involved in energy production pathways such as OXPHOS and TCA cycle, which are downregulated in aging MuSCs. 43 The promising effects of NR supplementation were confirmed in a clinical trial involving 20 BMI-discordant monozygotic twin pairs. One twin received oral NR (250–1000 mg/day) for 5 months, while the other received a placebo. NR supplementation resulted in a ∼14% increase in the number of mitochondria and upregulated the expression of genes involved in mitochondrial biogenesis (SIRT1/ERRα/TFAM/MFN2) in the NR-treated twin. Regarding MuSCs, NR supplementation reduced Pax7 and increased MyoD expression, suggesting that NR in humans may partially restore the activation and differentiation deficit of human myoblasts. Interestingly, NR-treated twins also showed an increased proportion of Faecalibacterium prausnitzii in their microbiota—a bacterium capable of synthesizing NR—suggesting the microbiota may play a role in fulfilling the body’s NAD+ pool demands. 116 Altogether, further human studies are needed to fully understand the therapeutic benefits of NAD supplements as well as extensive studies of the critical enzymes that utilize NAD+ as a cofactor to mediate its beneficial effects on elderly or diseased human patients.
In addition to a healthy diet, a growing body of evidence highlights caloric restriction (CR) as a promising intervention for delaying age-associated muscle dysfunction. 117 –119 In a study where 2- and 18-month-old mice underwent a 12-week CR regimen, both age groups exhibited a significant increase in MuSC numbers, enhanced myogenic colony-forming capacity ex vivo, and improved MuSC engraftment and muscle regeneration in mdx recipient mice following transplantation. Notably, CR was also associated with increased mitochondrial mass and improved mitochondrial function, promoting a metabolic shift toward OXPHOS over glycolysis, an adaptation particularly beneficial in aged muscle. 118 The mechanisms by which CR delays aging are still being elucidated, but one proposed pathway involves the inhibition of the nutrient-sensing mechanistic target of rapamycin (mTOR) and the activation of autophagy. 120 This is particularly relevant in aging, as mTOR signaling becomes chronically elevated in aged skeletal muscle, contributing to cellular senescence and stem cell exhaustion. 121,122 Interestingly, pharmacological inhibition of mTOR using rapamycin has been shown to extend lifespan across multiple species, including yeast, worms, flies, and mice. 120,123 –125 These findings raise the possibility of using CR and mTOR inhibition, individually or in combination, as a new strategy to delay aging and preserve stem cell function in humans.
TF reprogramming
Another strategy being explored to rejuvenate aged MuSCs is based on re-expressing four “reprogramming” Yamanaka factors (OKSM). 126 The expression of the OCT4, KLF4, SOX2, and MYC (OKSM) in differentiated cells has been reported to induce pluripotency and reverse several features of aging. 127 Controlled reprogramming is essential, as continuous OKSM expression has been associated with teratoma formation. 128 The rejuvenating effects of partial OKSM induction in reversing aging in skeletal muscle were demonstrated using mice carrying a cassette encoding for OKSM under the control of the Col1a1 promoter, which allows the expression of OKSM in multiple cell types, including muscle fibers and MuSCs after doxycycline treatment. In 12-month-old adult mice, intramuscular doxycycline injections were given to the TA muscles for 3 weeks, followed by CTX-induced muscle injury. This led to improved muscle regeneration 10 days postinjury, marked by increased CSA of the injured fibers and an increase in the number of Pax7+ve MuSCs. 129 The beneficial effects of OKSM expression were also demonstrated in another work where the reprogramming occurred only in muscle fibers after doxycycline treatment. 130 Specifically, mice treated with doxycycline for 3 weeks prior to muscle injury displayed improved regeneration, with increased CSA of muscle fibers 7 days post-CTX injury. Additionally, inducing OKSM expression in myofibers more effectively activated MuSCs in quiescent aged muscles, as shown by increased in vivo BrdU incorporation and MyoD expression. This enhanced activation was linked to upregulated p21 in muscle fibers, leading to the downregulation of Wnt4, which in turn promoted MuSC activation and proliferation. 130 While spontaneous MuSC activation in uninjured muscle tissue could deplete the stem cell pool, this strategy offers the advantage of controlled OKSM induction, which can be triggered or blocked through doxycycline treatment.
The beneficial impact of transient OKSM expression was also demonstrated in MuSCs in vitro, where MuSCs isolated from both young and aged mice were transfected with mRNAs expressing OKSM, along with LIN28 (which facilitates OCT4 expression). Interestingly, OKSM induction in aged MuSCs resulted in augmented activation and differentiation, as shown by increased EdU incorporation, higher myogenic fusion index in differentiating myosin heavy chain positive (MyHC+ve) myoblasts, and increased mitochondrial mass. 131 While the precise mechanisms behind OKSM’s rejuvenating effects remain unclear, studies have suggested that short-term OKSM induction in fibroblasts from mice with premature aging reduced DNA damage (as indicated by lower γ-H2AX foci and 53BP1 expression), downregulated senescence markers (p16INK4a and p21CIP1), and restored youthful levels of histone modifications (H3K9me3 and H4K20me3 which are downregulated and upregulated during aging, respectively). 129 It would be interesting to explore whether aged MuSCs exposed to partial OKSM would exhibit the same reversal of these aging hallmarks.
Pharmacological intervention
Pharmacological interventions aiming at rejuvenating MuSCs through the modulation of their activation have emerged as a promising avenue to combat aged-associated muscle decline. For example, inhibition of the p38 MAPK pathway was sufficient to rescue the defects in proliferation (increased cell number after 7 days in culture on hydrogel) and asymmetrical division ex vivo and prevented the upregulation of the senescence marker p16INK4a in vivo in aged MuSCs in culture. 132,133 Strikingly, p38 MAPK inhibition enhanced the engraftment of aged MuSCs by more than 50% when transplanted into recipient mice. 133 Similarly, targeted inhibition of the JAK/STAT pathway, specifically JAK2 and STAT3, increased proliferation in myofiber-associated MuSCs from 18 months old mice (i.e., higher number of Pax7+ve cells/myofiber), ameliorated muscle regeneration in 24 months old mice (i.e., increased CSA at 5 days postinjury) and improved aged MuSC engraftment after transplantation into recipient mice. 26,27
In aging muscles, upregulated canonical Wnt signaling is associated with acquiring a more fibrotic phenotype in MuSCs, 24 which is further exacerbated by a reciprocal interaction with TGF-β signaling. 134,135 To counter this, the use of Wnt inhibitors has been proposed as a strategy to restore the myogenic potential of aged MuSCs, as it has been shown to increase their proliferation in vivo and reduce muscle fibrosis, possibly through the inhibition of their fibrogenic conversion. 24 Additionally, treatment with a combination of oxytocin and TGF-β inhibitors has yielded positive effects on the muscle tissue of aged mice, including an increase in newly formed muscle fibers, a reduction in interstitial fibrotic tissue, and lower levels of the p16INK4a senescence marker protein in mononucleated cells, measured 5 days post-CTX injury. 25 Another pharmacological strategy consists of the systemic targeting of chronic inflammation, which is a hallmark of aging, via inhibition of NF-κB activation in muscle fibers of aged mice. This approach has been shown to increase the CSA of muscle fibers 7 days postcryoinjury and improve colony formation efficiency of MuSCs ex vivo. 136
Other potential interventions have focused on activating pathways typically associated with vertebrate longevity. The composition of extracellular matrix (ECM) deposition in the MuSC niche is crucial in determining tissue stiffness, which in turn influences MuSC fate. 137 MuSCs preferentially adhere to fibronectin, which interacts with beta1-integrin (ITGβ1) on their cell surface and downregulates p38 MAPK activity. Interestingly, aging leads to reduced fibronectin levels, and administration of fibronectin has been shown to restore MuSC activation defects specifically in aged muscles. 138 Importantly, aged MuSCs isolated from mice treated with fibronectin exhibited increased proliferation and myogenic commitment in vitro, as shown by elevated Ki67 and MyoD protein levels, and 7 days following glycerol-induced injury, fibronectin treatment accelerated muscle regeneration. 138 In another example, treatment with a β1-integrin activating antibody enhanced muscle regeneration in vivo, as measured by an increased number of eMyHC+ve myofibers 3 days post-CTX injury. β1-integrin activation also promoted the expansion of the Pax7+ve fraction of the stem cell pool. 139
Lastly, MuSC activation relies on autophagy to supply rapid energy needs. Pharmacological inhibition of autophagy was shown to disrupt mitochondrial function, reduce ATP production, and trigger senescence. 35,56 While young MuSCs use autophagy efficiently, aged MuSCs are more prone to fail to initiate autophagy and instead undergo apoptosis. 140 As a counter mechanism, the 5′ AMP-activated protein kinase (AMPK) pathway plays a critical role in MuSC fate decisions when autophagy cannot provide adequate energy. It has been reported that reduced AMPK/p27Kip1 activation in aged MuSCs impairs proliferation and survival, but these defects can be reversed by pharmacological activation of the AMPK pathway. 141 Altogether, these data highlight multiple pharmacological agents that will need further investigations in human studies to see whether they can reverse the negative effects of aging on the muscle system.
Conclusions and Future Perspectives
Recent advancements in understanding the complex molecular mechanisms driving age-associated muscle dysfunction have provided new insights into potential therapeutic strategies for rejuvenating aged MuSCs (Fig. 1). These strategies primarily focus on modulating intrinsic pathways within MuSCs or restoring the supportive environment of their niche (Table 1). However, further research is needed to develop combinatory therapies that address both MuSC dysfunction and niche-related impairments. We suspect that further research in mice and humans will be essential for translating these basic and preclinical studies into human therapies to counteract age-related muscle loss and improve muscle health in the elderly.
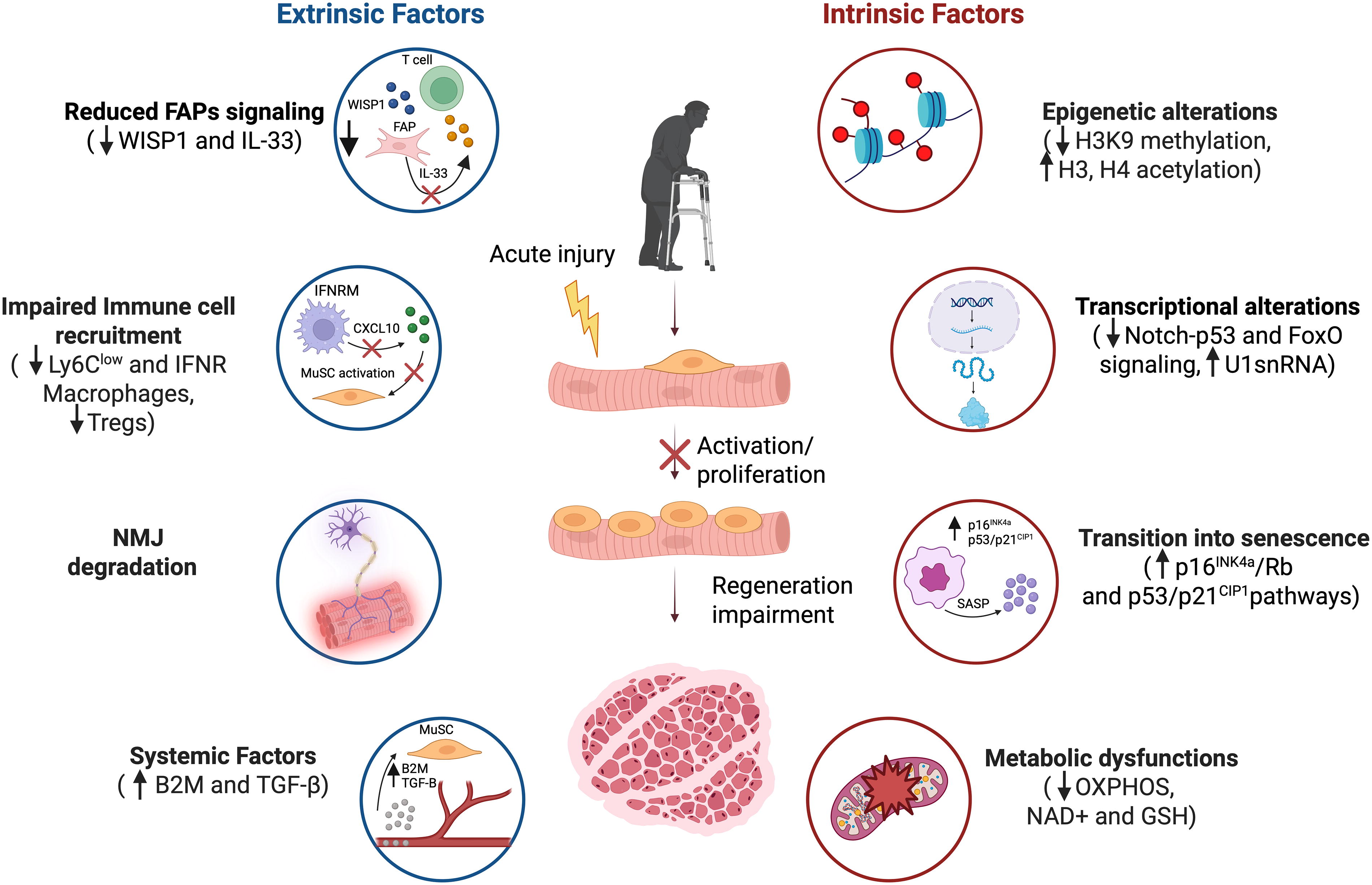
Overview of intrinsic and extrinsic mechanisms regulating MuSC aging. During aging, MuSCs undergo intrinsic, cell-autonomous changes, including epigenetic alterations (e.g., decreased H3K4 methylation and increased acetylation of histones H3 and H4), dysregulation of key signaling pathways (such as Notch-p53 and FoxO), and upregulation of the U1 spliceosomal RNA (U1snRNA), persistence in a senescent state following injury, and accumulation of metabolic dysfunctions (imbalance between glycolysis and OXPHOS, reduction of NAD+, and GSH availability). These intrinsic alterations are shaped by extrinsic, noncell-autonomous factors. Cells within the immediate niche, including FAPs and immune cells, modulate MuSC behavior by secreting cytokines and growth factors that act directly on MuSCs (e.g., CXCL10 from IFN-γ-activated macrophages, WISP1 from FAPs) or regulate recruitment of additional cells to the injury site (e.g., IL-33 from FAPs). MuSCs are also influenced by NMJ interactions, with NMJ integrity and regeneration impacting MuSC function. Additionally, systemic factors enriched in aged blood (such as B2M and TGF-β) have detrimental effects on MuSC function. B2M, beta-2-microglobulin; FAPs, fibro-adipogenic progenitors; FoxO, Forkhead box O; GSH, reduced glutathione; IFN-γ, interferon-gamma; IL-33, interleukin-33; NAD+, nicotinamide adenine dinucleotide; NMJs, neuromuscular junctions; OXPHOS, oxidative phosphorylation; TGF-β, transforming growth factor beta; U1 snRNA, U1 small nuclear RNA; WISP1, Wnt1-inducible signaling pathway protein-1.
Footnotes
Acknowledgments
The authors thank members of the Almada Lab for their critical reading of this article. Figure 1 was created using ![]() .
.
Authors’ Contributions
Conceptualization: M.L. and A.E.A. Writing—original draft, review and editing: M.L. and A.E.A. Final draft and editing: M.L. and A.E.A.
Author Disclosure Statement
The authors declare no competing financial interests.
Funding Information
This work was supported by an NIH R01 AR080753 to A.E.A.