
Abstract
As a typical E3 ligase, tripartite motif-containing 65 (TRIM65), is implicated in the modulation of biological processes, such as metastasis, proliferation, and apoptosis. However, the function of TRIM65 in prostate cancer (PCa) and its potential mechanism have not yet been excavated. In this work, we affirmed Tripartite motif-containing protein 65 (TRIM65) as a new oncogene in PCa, which accelerated PCa cell proliferation and impeded cell ferroptosis. In vivo, depletion of TRIM65 inhibited PCa tumorigenesis and metastasis. Mechanically, our findings uncovered that TRIM65 enhances NKD inhibitor of WNT signaling pathway 2 (NKD2) degradation via the ubiquitin-proteasome signaling. TRIM65 facilitated proliferation and restricted ferroptosis via downregulating NKD2 levels. Moreover, TRIM65 activated the wingless-integrated/β-catenin pathway in PCa cells via inhibiting NKD2. Taken together, these data uncovered that TRIM65 controls PCa proliferation, and ferroptosis and regulates the Wnt/β-catenin signaling via directly targeting NKD2 for ubiquitination degradation. Our study provides insights into the multifaceted regulatory role of TRIM65 in the development of PCa, laying the foundation for exploring new therapeutic approaches.
Introduction
Prostate cancer (PCa) is among the most common cancers globally and the fifth leading cause of cancer-related deaths in men. 1,2 PCa is often challenging to detect in its early, dormant stages, leading to most patients being diagnosed in the middle or late stages. 3,4 In recent years, numerous therapeutic strategies for PCa, including chemotherapy, androgen deprivation therapy (ADT), and targeted therapy for DNA repair, have been extensively explored. 5 Nevertheless, These approaches often show limited efficacy, as most patients with advanced PCa eventually develop resistance to castration and drugs, leading to cancer-related death. 6 –8 This stark reality highlights the urgent need for innovative therapies and a deeper understanding of the molecular mechanisms behind PCa progression.
Ubiquitin-mediated protein degradation pathways are crucial for eliminating transient regulatory proteins implicated in DNA repair, morphogenesis, cell cycle regulation, protein quality control, and more. 9 Posttranslational modifications, including the ubiquitin-proteasome system, modulate nearly all tumor suppressors and oncogenes. 10 Thus, identifying and targeting E3 ligases involved in the modulation of tumor suppressor and oncoprotein proteins has become a primary focus of cancer research. Recent research has uncovered that E3 ubiquitin ligases, such as carboxyl terminus of hsc-70-interacting protein (CHIP), are implicated in the occurrence of PCa. Oncogenic factor CHIP accelerated proliferation and inhibited apoptosis via regulating the AKT signaling pathway. 11 E3 ubiquitin ligase NEDD4L restrained cell proliferation by regulating PHF8 via the ubiquitin-proteasome signaling in PCa. 12 Numerous research has confirmed that members of the E3 ligase tripartite motif (TRIM) family of proteins also play a role in regulating PCa. TRIM47 depletion restricted the malignant behaviors of PCa cells by modulating the MDM2/p53 pathway. 13 TRIM36 impeded neuroendocrine differentiation of PCa through hexokinase 2 (HK2) ubiquitination and glutathione peroxidase 4 (GPX4) knockdown. 14 TRIM65 accelerated renal cell carcinoma by ubiquitinating and degrading BTG anti-proliferation factor 3 (BTG3). 15 However, whether TRIM65 is involved in PCa progression is largely unknown.
Here, we verified TRIM65 as a novel E3 ubiquitin ligase for NKD2. NKD2, part of the mammalian homologous family of naked cuticle proteins, is located on chromosome 5p15.3. 16 NKD2 has been reported to antagonize typical Wnt signals. Dong et al. unveiled that epigenetic silencing of NKD2, a key component of Wnt signaling, expedites breast cancer growth. 17 Cao et al. elaborated that depletion of NKD2 facilitates esophageal cancer development by activating the Wnt pathway. 18 However, whether TRIM65 degrades NKD2 by ubiquitination and then activates the Wnt pathway to affect PC progression is unknown.
In our work, we observed a high level of TRIM65 in PCa tissues and cells. Depletion of TRIM65 could distinctly restrict cell proliferation while TRIM65 silencing accelerated cell ferroptosis. Further, TRIM65 modulated PCa cell proliferation and ferroptosis mainly through direct ubiquitination with NKD2 and activation of Wnt signaling. These data indicated that TRIM65 acts as a novel oncogene by promoting the ubiquitination and degradation of NKD2.
Materials and Methods
Clinical samples
Forty pairs of PCa tissues and adjacent normal tissues were collected from PCa patients admitted at The First Affiliated Hospital of Jinzhou Medical University. Informed consent was obtained from each patient enrolled.
Cell culture and transfection
PCa cell lines DU145, PC-3, Lymph Node Carcinoma of the Prostate (LNCaP), and 22RV1 and normal human prostate epithelial cell line (RWPE-1) were purchased from the American Typical Culture Collection Center (ATCC, Manassas, VA, USA). Cells were cultured in PRMI-1640 or Dulbecco's Modified Eagle Medium (DMEM) media, each supplemented with 10% fetal bovine serum [(FBS), PAN Biotech, cat#ST30-3302] and antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin).
The short hairpin RNA vectors targeting TRIM65 (sh-TRIM65), negative control (sh-NC), TRIM65 and NKD2 overexpression plasmid (oe-TRIM65 and oe-NKD2) and the NC plasmid (an empty vector, oe-NC) were purchased from GenePharma (Shanghai, China). Overexpressing plasmid (2 μg) or shRNA (1.5 μg) of indicated genes were transfected into cells using Lipofectamine 3000 (Invitrogen) following the manufacturer’s protocols. After 48 hours, cells were collected for subsequent experiments.
Cell counting kit-8 (CCK-8) assay
Cells were seeded into each of 96-well plates (5 × 103/well). After cultured for the indicated time, CCK-8 reagent was added (20 μL/well, Cat# 40203ES76, Yeasen, Shanghai, China). After incubation for 3 hours at 37°C, the optical density was recorded at 450 nm using an automatic microplate reader.
Colony formation assay
A colony colony-forming assay was performed to determine the clonogenic activity of PCa cells. Briefly, PCa cells were cultured in 6-well plates until colonies became visible to the naked eye. The colonies were subsequently rinsed with PBS, fixed with methanol, and stained with Giemsa.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells or tissues using TRIzol reagent (Invitrogen). Reverse transcription was performed using the PrimeScript RT reagent kit (TaKaRa), and qPCR was conducted using SYBR green master mix on a real-time PCR system. Gene levels were normalized to GAPDH, and relative levels were assessed using the 2-ΔΔCT method. The primers used in this study were as follows: TRIM65, Forward 5′-TGAGAGCCAGCCTGGAGGTTAC-3′ and Reverse 5′-TGTAGCAGGCTGCTGAACTTGC-3′; NKD2, Forward 5′-GACAACTCCTCAGCGCAGATGA-3′ and Reverse 5′-GTCATAGAGCGTGAACGTCCAC-3′; GAPDH, Forward 5′-GTCTCCTCTGACTTCAACAGCG-3′ and Reverse 5′-GTCTCCTCTGACTTCAACAGCG-3′.
Western blot
Cells were harvested, lysed using ice-cold lysis buffer, and then quantified with a BCA protein assay kit (Beyotime, P0012, China). Protein was separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a Polyvinylidene Fluoride (PVDF) membrane, and incubated with Tris-Buffered Saline with Tween-20 (TBST) blocking solution. The PVDF membrane was then incubated with primary antibodies: SLC7A11 (CSB-PA145393, 1:500, CUSABIO), GPX (29329–1-AP, 1:2000, Proteintech), TRIM65 (ab154821, 1/1000, Abcam), NKD2 (16699–1-AP, 1:500, Proteintech), MYC (#13987, 1:1000, Cell signaling), Cyclin D1 (#2978, 1:1000, Cell signaling), β-catenin (#8480, 1:1000, Abcam), and GAPDH (#2118, 1:1000, Cell signaling) at 4°C overnight, followed by incubation with a horseradish peroxidase-conjugated secondary antibody (A0208, 1:3000, Beyotime) at room temperature for about 1 hours. Finally, an enhanced ECL chemiluminescence kit was used to prepare the membrane, and the signal bands were quantified by ImageJ software.
Co-immunoprecipitation (Co-IP)
Total protein was extracted using RIPA lysis buffer (Beyotime, P0013B, China) supplemented with protease inhibitor cocktails (Beyotime, ST506, China). The lysates were centrifuged to obtain the supernatant. Proteins (1000 μg) were incubated with 2 μg of primary antibodies against TRIM65 (ab154821, 1/1000, Abcam) or NKD2 (16699-1-AP, 1:500, Proteintech) overnight at 4°C. Protein-antibody complexes were collected from lysates using Protein G-agarose beads (Santa Cruz Biotechnology, CA, USA) for 6 hours at 4°C. The agarose-bound immunoprecipitated complexes were centrifuged, washed, and assessed by western blot assay.
Ubiquitination detection assay
Cells were lysed and boiled (10 minutes). Samples were sonicated and incubated with dilution buffer for 30 minutes. Samples were incubated with anti-NKD2 (16699–1-AP, 1:500, Proteintech) antibody followed by incubation with protein A/G IP magnetic beads for 12 hours. NKD2 ubiquitination was detected using a western blot with an anti-ubiquitin antibody (Abcam, 1:1000, ab140601).
Cycloheximide (CHX) assay
To assess protein degradation rates, cells were treated with the protein synthesis inhibitor (CHX; C7698, Sigma, 100 µg/mL). Cells were harvested at various time points (0, 2, 4, 6, and 8 hours) after CHX treatment. Total protein was extracted, and the levels of target proteins were analyzed by western blotting.
The detection of glutathione (GSH), Fe2+, and Malondialdehyde (MDA) content
The evaluation of GSH, Fe2+, and MDA content, GSH detection kit (cat. no. A006-2–1, Nanjing Jiancheng Bioengineering Institute), iron colorimetric assay kit (cat. no. E-BC-K881-M; Elabscience Biotechnology, Inc.), and MDA detection kit (cat. no. A003-1, Nanjing Jiancheng Bioengineering Institute) were used following the instructions provided by the manufacturer.
Reactive Oxygen Species (ROS) detection
The prepared 2ʹ,7ʹ-dichlorofluorescin diacetate (DCFH-DA) diluent was added to cells seeded in 6-well plates and incubated for 30 minutes at 37°C in the dark. The cells were then washed three times with PBS, digested with trypsin, and collected by centrifugation. The cell pellets were resuspended in PBS. ROS expressions were measured using a flow cytometer (Beckman Coulter, Indianapolis, IN, USA).
Animal study
Three-week-old male BALB/c nude mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. For the xenograft mice model, PC-3 cells infected with lentivirus vectors (sh-NC or sh-TRIM65) were injected subcutaneously in the dorsal flank of each mouse. The tumor dimensions (length and width) were measured with a caliper every 5 days. Xenograft growth was tracked for 6 weeks postinjection. All mice were euthanized to collect fresh tumors, and their weights were recorded. For the lung metastasis model, the above cells were injected into the tail vein of each mouse. After another 8 weeks of growth, the fluorescence intensity of the tumor was assessed by Living Image software. All the experiment protocol was approved by the Ethics Committee of The First Affiliated Hospital of Jinzhou Medical University (HJMU02061, date of approval: 23, March 2023).
Immunohistochemistry (IHC)
The sections were dewaxed with xylene, dehydrated with ethanol, and boiled in citrate for 10 minutes. The sections were then incubated with 3% H2O2 at room temperature for 10 minutes. Sections were blocked with goat serum and incubated with primary antibodies Ki-67 (ab15580, Abcam) or Cyclin-D1 (ab134175, Abcam) overnight at 4°C. The next day, sections were washed three times with phosphate-buffered saline with Tween-20 (PBST) and incubated with a secondary antibody for 30 minutes at room temperature. Finally, sections were stained with DAB and hematoxylin, and images were captured using Olympus X71 inverted microscope (Olympus Corporation, Tokyo, Japan).
Statistical analyses
All the statistical analyses were performed using GraphPad Prism 6.0 (GraphPad, USA). All data were presented as means ± SD. Statistical differences between groups were analyzed using Student’s t-test or one-way ANOVA. p < 0.05 was regarded as statistical significance.
Results
TRIM65 level was enhanced in PCa tissues and cells
The University of Alabama at Birmingham Cancer Data Analysis portal (UALCAN) database was used to assess TRIM65 levels in prostate adenocarcinoma (PRAD) clinical samples. The results displayed that TRIM65 expression was enhanced in PRAD tissues (Fig. 1A). Similarly, RT-qPCR and western blot revealed that TRIM65 expression was aberrantly higher in PCa tissues and cell lines (Fig. 1B–D). Moreover, the TRIM65 level gradually enhanced with the Gleason score, and the TRIM65 level was distinctly higher in PRAD patients with lymph node metastasis (Fig. 1E and F). As shown in Figure 1G, the overall survival rate of the high TRIM65 expression group was lower than that of the low TRIM65 expression group. Taken together, TRIM65 expression was distinctly elevated in PCa and linked to poor survival.
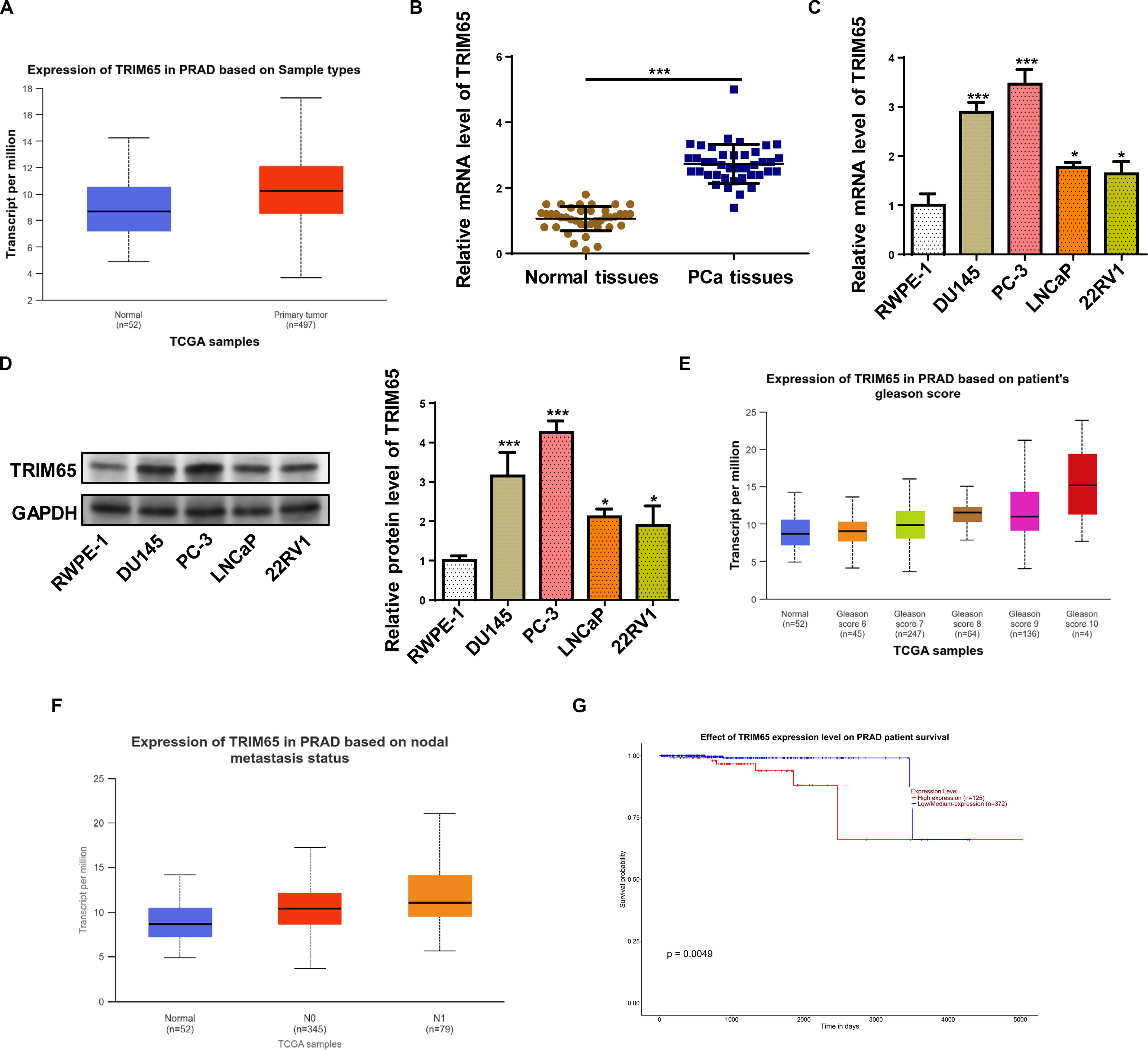
TRIM65 expression was upregulated in PCa tissues and cells.
Depletion of TRIM65 suppressed the proliferation and metastasis of PCa cells
Due to the high level of TRIM65 in PCa cells, we silenced TRIM65 in these two cell lines to verify its biological function (Fig. 2A and B). Next, the transfected cells were subjected to CCK-8 and colony formation assays. As demonstrated in Figures 2C and D, TRIM65 depletion substantially restrained the viability and number of colonies of DU-145 and PC-3 cells. Moreover, xenograft tumors were established using PC-3 cells infected with sh-NC or sh-TRIM65. Consistent with in vitro results, TRIM65 depletion successfully inhibited tumor growth, as shown by tumor size, weight, and volume (Fig. 2E–G). Moreover, an IHC assay was utilized to detect pivotal cell proliferation markers (Ki67 and Cyclin-D1). The results indicated that Ki67 and Cyclin-D1 levels declined in tumors formed by PC-3-sh-TRIM65 cells (Fig. 2H). Further, to explore the connection between TRIM65 and the metastatic ability in vivo, PC-3 cells transfected with sh-TRIM65 were injected into BALB/c-nu mice via the tail vein. In the pulmonary metastasis model, TRIM65 knockdown distinctly impeded lung metastasis of PC-3 cells (Fig. 2I–J). In sum, inhibition of TRIM65 impeded tumor growth and metastasis both in vitro and in vivo.
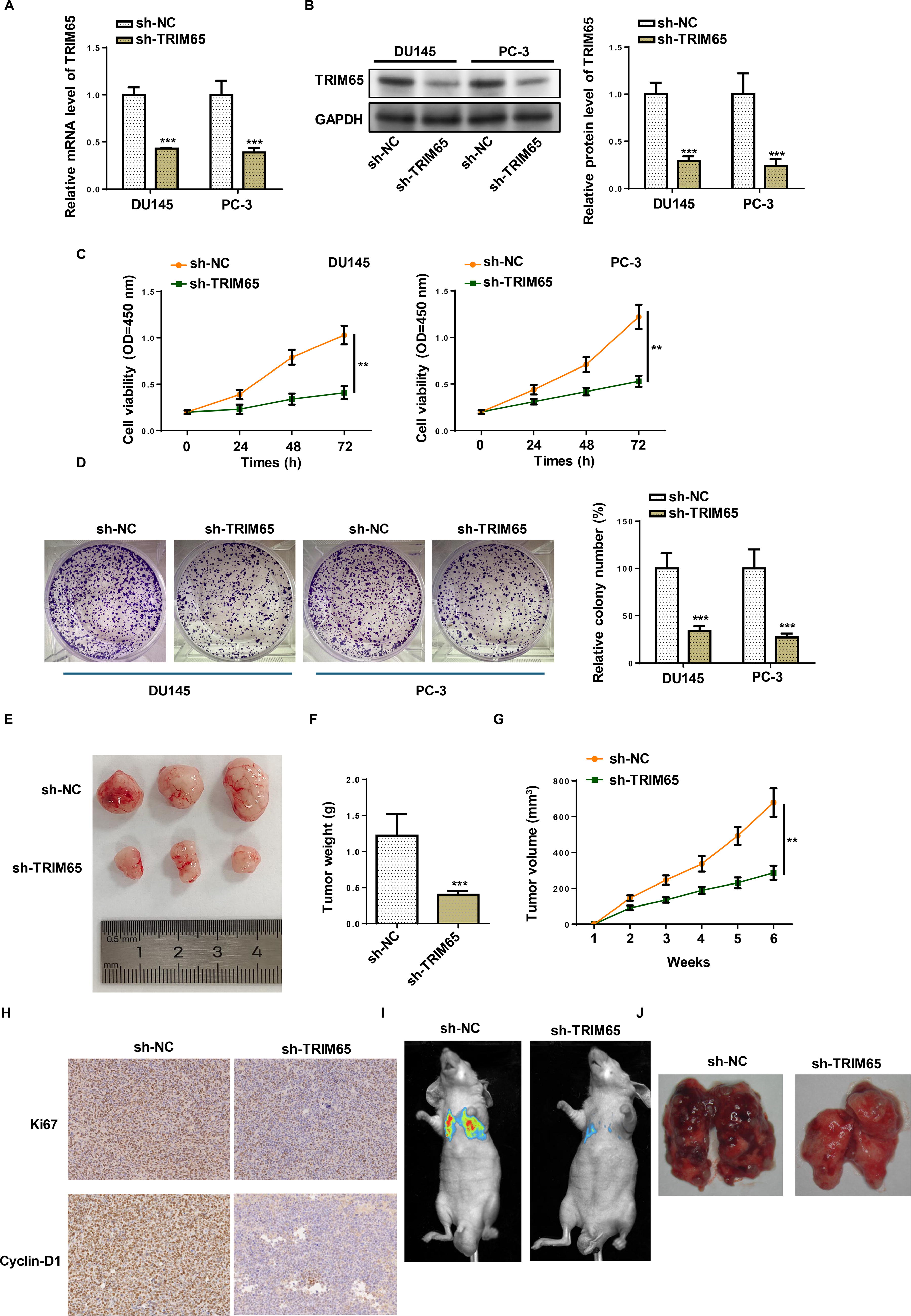
Depletion of TRIM65 inhibited the proliferation and metastasis of PCa cells.
Depletion of TRIM65 promoted ferroptosis in PCa cells
Next, we investigated whether TRIM65 could affect ferroptosis in PCa cells. Intracellular ROS of PCa cells with TRIM65 depletion was stained by DCFH-DA and assessed by flow cytometry assay. As exhibited in Figure 3A, the ROS level was prominently enhanced by TRIM65 silencing in PCa cells. Moreover, TRIM65 knockdown promoted MDA level and Fe2+ content and inhibited GSH levels in DU-145 and PC-3 cells (Fig. 3B–D). Further, TRIM65 knockdown declined SLC7A11 and GPX4 protein levels in PCa cells (Fig. 3E). These results implied that TRIM65 impedes ferroptosis in PCa cells.
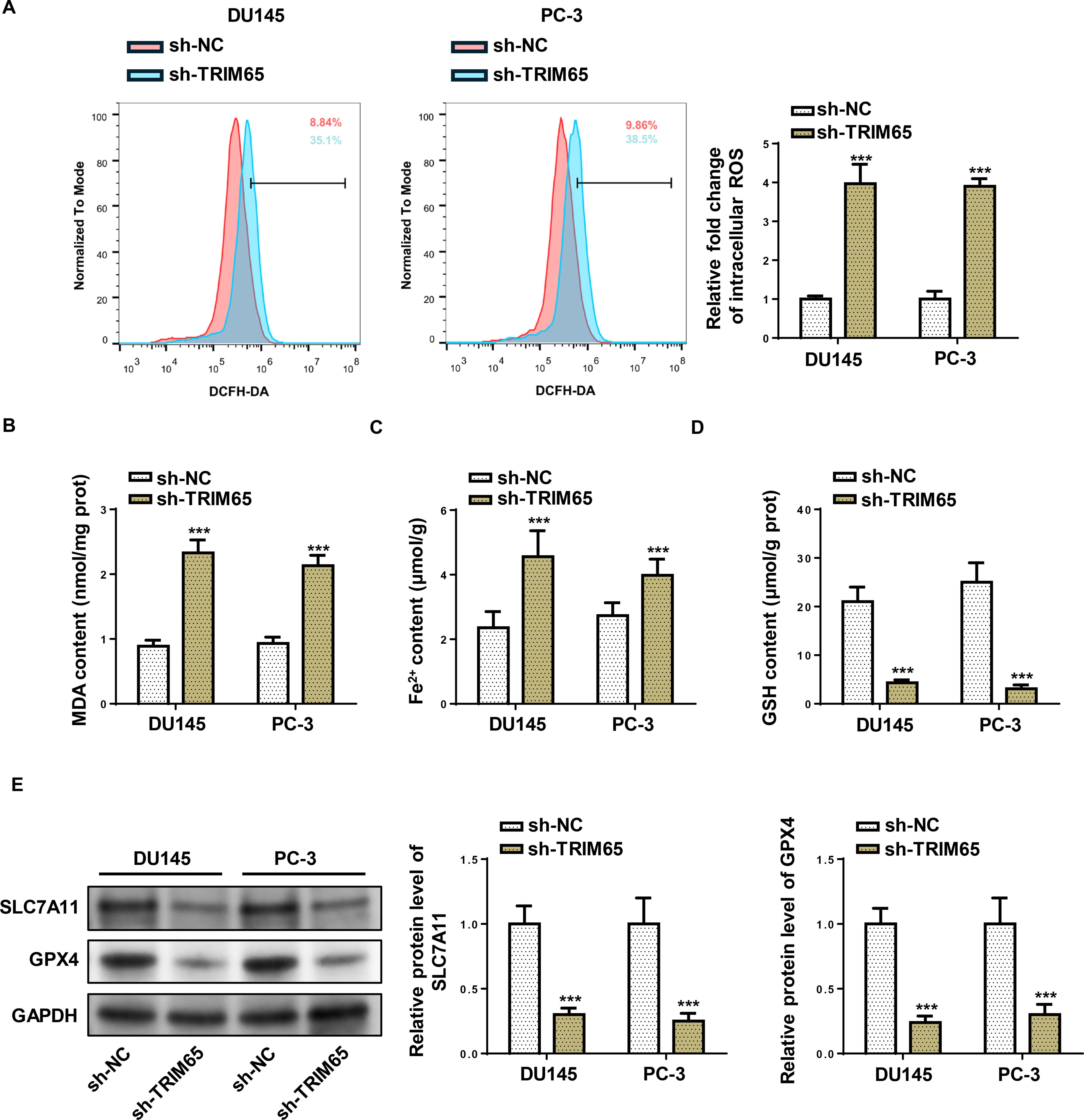
Depletion of TRIM65 promoted ferroptosis in PCa cells.
TRIM65 degraded NKD2 via ubiquitination
NKD2 has been revealed to suppress the development of various cancers. 19,20 UALCAN database demonstrated that NKD2 declined in PRAD tissues (Fig. 4A). Moreover, analysis of the UALCAN database confirmed low expressions of NKD2 correlated with a low survival rate of PRAD patients (Fig. 4B). RT-qPCR and western blot further revealed that NKD2 level was aberrantly lower in PCa tissues and cell lines (Fig. 4C and D). Moreover, we found TRIM65 led to a downregulation of NKD2 protein level but not the mRNA level in PCa cell lines (Fig. 4E and F) implying that TRIM65 might modulate NKD2 level at protein level. Then the interaction between TRIM65 and NKD2 was probed. Co-IP assay indicated that TRIM65 could bind to NKD2 (Fig. 4G). Besides, TRIM65-overexpressed PCa cells were treated with CHX, it was observed that the degradation of NKD2 was enhanced by TRIM65 addition with CHX treatment (Fig. 4H). Then we detected whether NKD2 was stabilized by TRIM65-regulated ubiquitination. Ubiquitination assay indicated that TRIM65 promoted NKD2 ubiquitination (Fig. 4I). These results hinted that TRIM65 could interact with NKD2 and promote its ubiquitination.
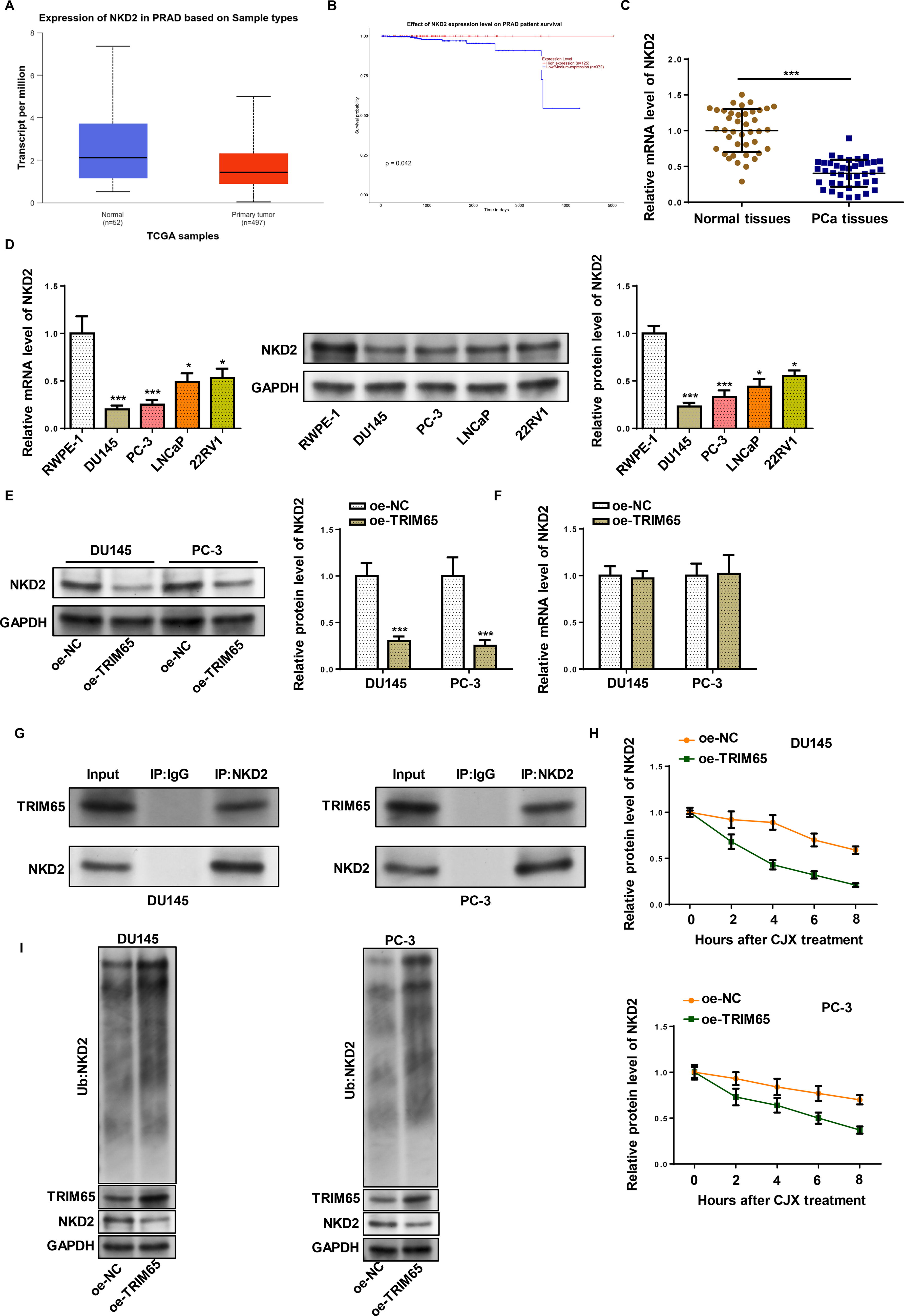
TRIM65 degraded NKD2 via ubiquitination.
NKD2 reversed the effects mediated by TRIM65 on PCa cell behaviors
To investigate whether TRIM65 regulates PCa progression by regulating NKD2, PCa cells were transfected with oe-TRIM65 alone or together with oe-NKD2. As exhibited in Figure 5A, the addition of TRIM65 inhibited NKD2 expression, which was abolished by NKD2 transfection. Notably, oe of NKD2 did not affect the level of TRIM65 in TRIM65-overexpressed PCa cells. Cell viability and number of colonies induced by TRIM65 were neutralized by NKD2 overexpression (Fig. 5B and C). Moreover, TRIM65 inhibited intracellular ROS, MDA, and Fe2+ levels and promoted GSH levels, which were neutralized after NKD2 addition (Fig. 5D–G). These findings indicated that TRIM65 accelerates cell proliferation and restrains ferroptosis by regulating NKD2.
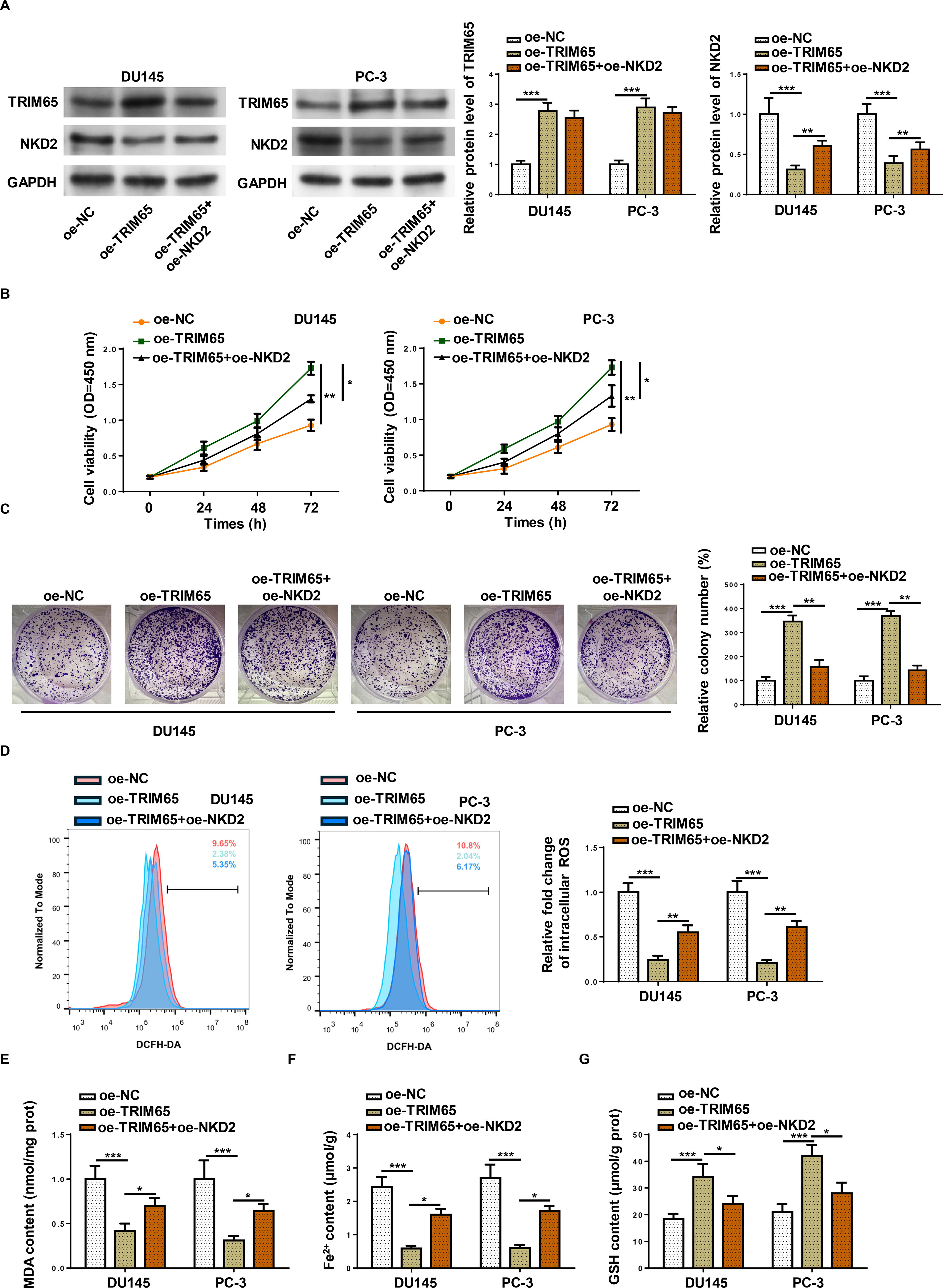
NKD2 reversed the effects mediated by TRIM65 on PCa cell behaviors. DU145 and PC-3 cells transfection with oe-NC, oe-TRIM65, and oe-TRIM65+oe- NKD2.
TRIM65 exerted oncogenic effects on PCa cells by activating the Wnt/β-catenin pathway via NKD2 downregulation
The Wnt/β-catenin pathway plays a critical role in ferroptosis, 21 and PCa progression. 22 Previous studies have demonstrated that NKD2 is a negative regulator of the Wnt/β-catenin pathway. 1 We examined whether TRIM65 was implicated in NKD2-mediated ferroptosis via Wnt/β-catenin signaling. MYC, Cyclin D1, and β-catenin protein levels were detected in PCa cells that were transfected with oe-TRIM65 with or without oe-NKD2. MYC, Cyclin D1, and β-catenin protein levels were upregulated in TRIM65-overexpressed PCa cells, which were abolished by NKD2 (Fig. 6A). In summary, the oncogenic role of TRIM65 in PCa is dependent on NKD2/Wnt/β-catenin axis.
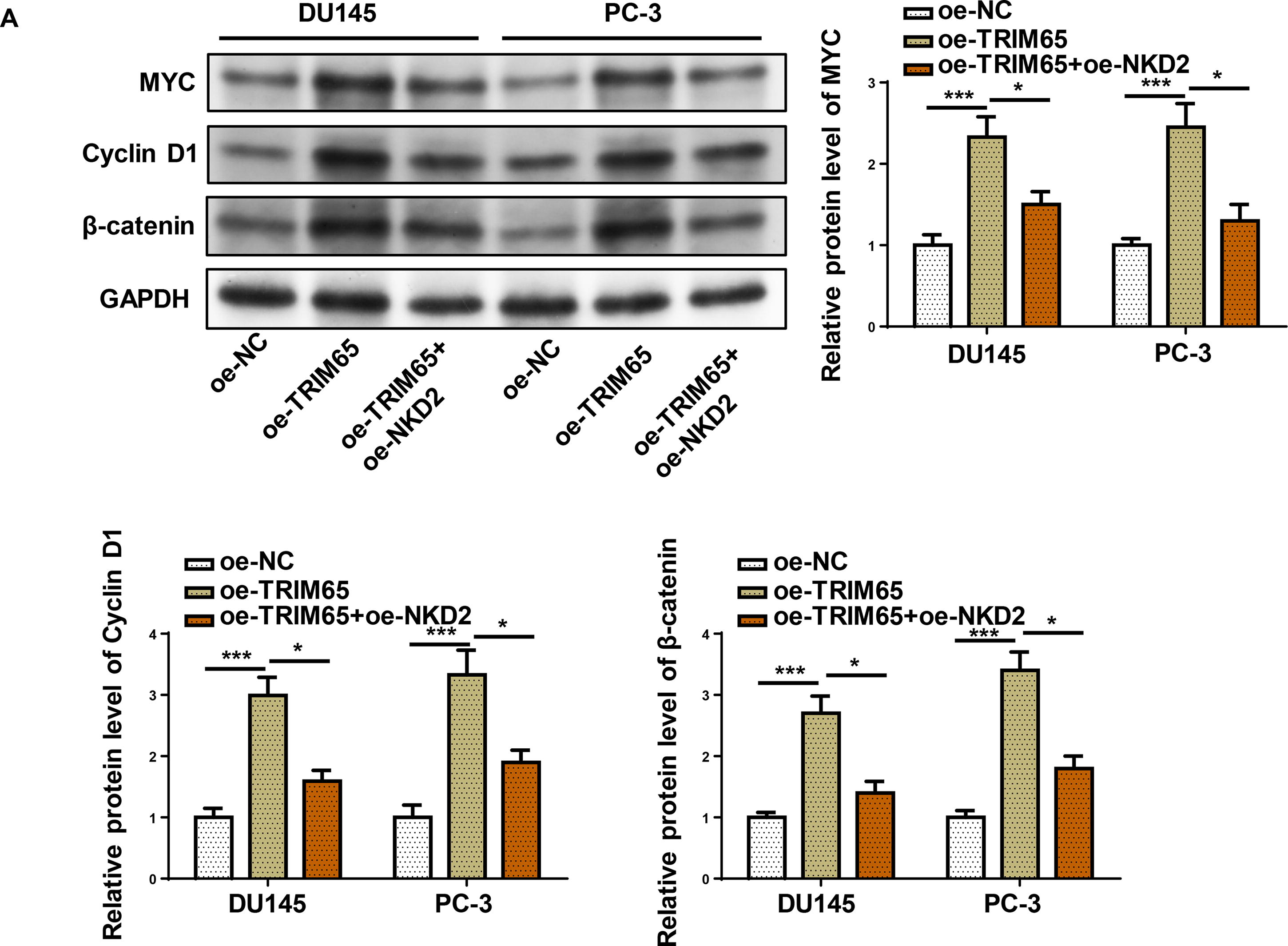
TRIM65 exerted oncogenic effects on PCa cells by activating the Wnt/β-catenin signaling pathway via NKD2 downregulation.
Discussion
In the past decade, there have been many advancements in PCa treatment, with ADT remaining the standard therapy for patients with metastatic PCa. 6 However, most patients eventually progress to a lethal stage of metastatic castration-resistant PCa. 7 Precision medicine aims to treat advanced cancers by affirming their specific genes. In this work, we reveal the expression and role of a new ubiquitination enzyme in the development of PCa. We demonstrated that TRIM65 degrades NKD2 protein by its ubiquitination, which subsequently activated the Wnt/β-catenin pathway, then inhibited cell proliferation and promoted ferroptosis in PCa (Supplementary Fig. S1).
As a known E3 ligase, TRIM65 regulates various cellular activities and related diseases primarily through the degradation of its specific substrates. TRIM65 has been uncovered to function as an oncogene in various tumor cancers, including pituitary tumors, 23 gastric cancer, 24 triple-negative breast cancer, 25 and cervical cancer. 26 Moreover, Xiong et al. indicated that RNF41, an E3 ubiquitin ligase, facilitates PCa metastasis by inducing actin remodeling. 27 Cheng et al. revealed that E3 ubiquitin ligase CHIP functions as an oncogene and activates the AKT signaling pathway in PCa. 11 However, no research to date has investigated the influences of TRIM65 on PCa. Consistent with TRIM65 levels in other cancer tissues and cell lines, we observed that TRIM65 was highly expressed in PCa tissues and cell lines. Knockdown of TRIM65 inhibited PCa cell proliferation. Moreover, by increasing the levels of ROS, MDA, and ferroptosis-related protein levels, and decreasing the levels of GSH-PX, it was proved that the loss of TRIM65 accelerated ferroptosis in PCa cells.
Ferroptosis is a newly identified form of programmed cell death marked by iron overload and lipid peroxidation accumulation. 28 It has been uncovered that ferroptosis suppresses tumor growth. 29 Cancer cells accumulate high levels of iron, unlike normal cells. 30 There are also significant differences in the sensitivity of different types of cancer cells to ferroptosis. 31 Several studies have demonstrated that Erastin can induce ferroptosis in PCa cells that harbor the RAS gene. Therefore, Erastin or RSL3, in combination with standard treatments, could impede the growth and migration of PCa cells in vitro and impede the development of advanced PCa in vivo. 30,32,33 The Wnt/β-catenin pathway plays a vital role in organ development, tissue homeostasis, and the pathogenesis of human diseases. Recently, ferroptosis has also been observed to be modulated by the Wnt/β-catenin signaling. Wang et al. elaborated that suppression of the Wnt/β-catenin signaling selectively enhances gastric cancer cell sensitivity to ferroptosis. 34 Moreover, according to another report, deficiency of the Wnt/beta-catenin pathway facilitates ferroptosis in melanoma. 21 In this study, we found that TRIM65 silencing promotes ferroptosis via inactivation of the Wnt/β-catenin pathway.
To further investigate the potential mechanism by which TRIM65 regulates the Wnt/β-catenin pathway and influences the progression of PCa, we focused on the Wnt/β-catenin pathway blocker NKD2. Previous research has revealed that NKD2 inhibits the development of many cancers. For instance, Jia et al. indicated that NKD2 impedes metastasis and invasion by regulating SOX18 in gastric cancer cells. 35 Cao et al. indicated that NKD2 restrains esophageal cancer development by suppressing the Wnt pathway both in vitro and in vivo. 18 In this work, we indicated for the first time that NKD2 is downregulated in PCa tissues and cell lines. Moreover, we observed that TRIM65 interacts with NKD2 and promotes its ubiquitination. TRIM65 accelerated cell proliferation and restrained ferroptosis by degrading NKD2. Furthermore, the addition of TRIM65 activated the Wnt/β-catenin pathway, which was reversed by NKD2. Anyway, we acknowledged some limitations in this study. Further studies will examine the effect of TRIM65 on other vital regulators in PCa progression. Whether some upstream signals participate in regulating the interaction between TRIM65 and NKD2 and affect the progression of PCa requires further research in the future.
In conclusion, the present work illustrated that TRIM65 exerts oncogenic effects on PCa by activating the Wnt/β-catenin pathway via degrading NKD2 by ubiquitination. Targeting the TRIM65/NKD2/Wnt/β-catenin axis could be an effective strategy for treating PCa.
Footnotes
Authors’ Contributions
C.W. and H.J.: Designed this study, performed all the experiments, analyzed the data, prepared the figures, and drafted the initial article. H.J.: Reviewed and revised the article. All authors read and approved the final article.
Ethics Approval and Consent to Participate
This study was approved by the Ethics Committee of The First Affiliated Hospital of Jinzhou Medical University.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.