
Abstract
Background:
Models of muscle cell culture allow simple manipulation to optimize gene transfer experiments, offering a significant opportunity to model muscle effects by allowing precise control of the cell culture environment in a cost-effective manner. Often, gene transfer is performed in myoblasts, which are then differentiated into myotubes, a skeletal muscle model. In this study, we aimed to compare the efficiency of direct transfection of differentiated myotubes by chemical and physical plasmid methods.
Methods:
Differentiated myotubes were transfected with reporter plasmids. Transfection efficiency and cell death were quantified. Proinflammatory proteins are produced by cells and tissues after DNA transfection; therefore, the secretion of proinflammatory chemokines was quantified.
Results:
In this study, we demonstrate that C2C12 myotubes can be effectively transfected using multiple methods. While a chemical method did not cause cell death, myotubes were not efficiently transfected. Electroporation delivery produced a higher transfection efficiency coupled with higher cell death. A subset of proinflammatory chemokine proteins was secreted, which replicated secretion by skeletal muscle.
Discussion:
These results suggest that differentiated myotubes can be directly transfected invitro, which may represent a useful experimental model. However, more studies are needed to assess the value of this model in predicting invivo responses.
Introduction
The three Rs in animal welfare are replacement, reduction, and refinement.1 When developing gene therapies, replacement with cells or models avoids the use of animals to the fullest extent possible. Undifferentiated cells in culture are often studied; however, differentiated cells may be a more appropriate model for tissues. Although there is no replacement for muscle invivo, the simple differentiation of myoblasts to myotubes can mimic aspects of skeletal muscle tissue invivo.2–4 In this study, experiments were performed in the C2C12 myotube model, an environment potentially more relevant to invivo studies than the myoblast. This model was used to compare nonviral gene transfer methods.
Several technologies produce efficient delivery of plasmid deoxyribonucleic acid (pDNA) transfection into cells and tissues, including chemical and physical methods.5 We compared direct transfection of C2C12 myotubes using two common methods, several chemical reagents, and electroporation (electrotransfer). This study first aimed to assess transfection efficiency and myotube survival.
Intramuscular injection of plasmid DNA is inflammatory.6 Intramuscular DNA electrotransfer is associated with the production of proinflammatory molecules7,8 and inflammation.9–11 These inflammatory effects may be due to the activation of pattern recognition receptor pathways, which are ubiquitously expressed in mammalian cells. In cells with functional signaling pathways, endosomal or cytosolic DNA sensors are activated by the introduction of exogenous nucleic acids inherent in DNA-based therapies.12 Therefore, a second aim was to evaluate the expression of proinflammatory chemokines induced by DNA transfection of myotubes to determine whether this expression was similar to that detected after invivo skeletal muscle delivery.
Materials and Methods
Cells and myotube formation
C2C12 mouse myoblast cells (CRL-1772, American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's Modified Eagle Medium (Corning, Thermo Fisher Scientific, USA) supplemented with 5% fetal bovine serum (Gibco, Waltham, MA, USA) and 1x Penicillin-Streptomycin (Gibco) in a 5% CO2 humidified incubator at 37°C. The cells regularly tested negative for mycoplasma infection using the Myco-Sniff Polymerase Chain Reaction Detection Kit (MP Biochemicals, Irvine, CA, USA). C2C12 myoblasts at 60-80% confluency were differentiated into myotubes by starvation for 8 days in medium containing 2% horse serum without antibiotics as previously described.13
pDNA transfection
Plasmids encoding Katushka fluorescent protein (pTurboFP635-C, Evrogen, Moscow, Russia) or luciferase (gWiz Luc, Aldevron, Fargo, ND, USA) driven by the cytomegalovirus-immediate early promoter/enhancer were commercially prepared (Aldevron, Fargo, ND, USA). Endotoxin levels were confirmed to be <100 endotoxin units/mg. Myoblasts and differentiated myotubes were transfected using several proprietary reagents, Transit LT1 (Mirus Bio LC, Madison, WI, USA), EndoFectin Max (GeneCopoeia, Rockville, MD, USA), and jetPrime (PolyPlus Transfection, New York, NY, USA) per the manufacturer's instructions. For electrotransfer, differentiated C2C12 myotubes in complete medium containing a final concentration of 0.25 µg/µL pDNA were electroporated using a BTX ECM 830 Square Wave Electroporator coupled with a Petri-Pulser electrode (PP35-2P, Harvard Apparatus, Holliston, MA, USA). A single 10 ms 160 V square wave pulse was delivered.14 Conditioned medium was collected after 48 h incubation in complete culture medium.
Luciferase Quantification
Four hours after transfection, culture medium was replaced with medium containing 150 µg/mL
Microscopic analysis
Crystal violet staining was performed as previously described15 and visualized microscopically. The number of myotubes was quantified using ImageJ.16 For Live–Dead staining, the medium was replaced 48 h after electrotransfer with two drops each of room temperature NucBlue® Live reagent and NucGreen® Dead reagent (ReadyProbes Cell Viability Imaging Kit, Thermo Fisher Scientific, Waltham, MA, USA) to 1 mL of medium.
Fluorescence was monitored microscopically (BZ-X700E, Keyence Corp., Itasca, IL, USA). Live cells were detected with a standard DAPI (4′,6-diamidino-2- phenylindole) blue filter set (excitation/emission maxima: 360/460 nm), and the nuclei of dead cells with compromised plasma membranes were detected with a standard fluorescein isothiocyanate/green fluorescent protein (green) filter set (excitation/emission maxima: 504/523 nm). A standard ET-mCherry, Texas Red® (Chroma Technology Corporation, Bellows Falls, VT), filter set (excitation/emission maxima: 560/630) was used to detect Katushka fluorescent protein expression. Fluorescence was quantified using the Hybrid Cell Count tool (Keyence). After loading the files in .jpg format into the software, we manually delimited the myotubes to specify the extraction area. The myotubes, defined as containing three or more live (NucBlue stained) nuclei, were tracked and counted using Image J software.12 Using the tool, the number of blue/live cells (NucBlue) and green/dead cells (NucGreen) within the delimited areas was then counted. For transfection efficiency, the red (Katushka expressing myotubes) were quantified and normalized to total viable myotubes. Assessors were blinded to the experimental groups until the completion of the study.
Chemokine quantification by bead array
Conditioned medium from electroporated myotubes was analyzed using a premixed multiplex panel (Mouse Cytokine/Chemokine Magnetic Luminex Assay, Millipore, Burlington, MA, USA) on a MAGPIX System (Luminex, Austin, TX, USA) per the manufacturer's instructions. All protein identifiers are per UniProt.17
Statistical analysis
Statistical evaluation of the differences between groups and graph preparation was conducted using GraphPad Prism 9.1.0 (San Diego, CA, USA). Since the data were normally distributed, significance was determined by a one-way ANOVA test followed by a Tukey-Kramer post-test. A p < 0.05 was considered statistically significant.
Results
Myoblast differentiation
Several culture conditions can be used to induce myotube formation from myoblasts.4 As observed previously, starvation of C2C12 myoblasts in 2% horse serum produced multinucleated myotubes13,15 (Fig.1).

Evaluation of C2C12 myotube differentiation. Representative brightfield images of crystal violet-stained cells. Myotube images were taken in sequence during differentiation. Images are shown on (a) day 0 and (b) day 8. Scale bar = 200 μm. (c) Quantification of 27 myotubes per group (****p < 0.0001).
pDNA delivery via chemical methods
Myoblast transfection was quantified after pDNA delivery using three commercial proprietary reagents (Fig.2). While each reagent significantly delivered plasmid DNA as denoted by luciferase transgene expression (p < 0.0001), jetPrime produced significantly higher expression than the other reagents or the plasmid only control (p < 0.0001).
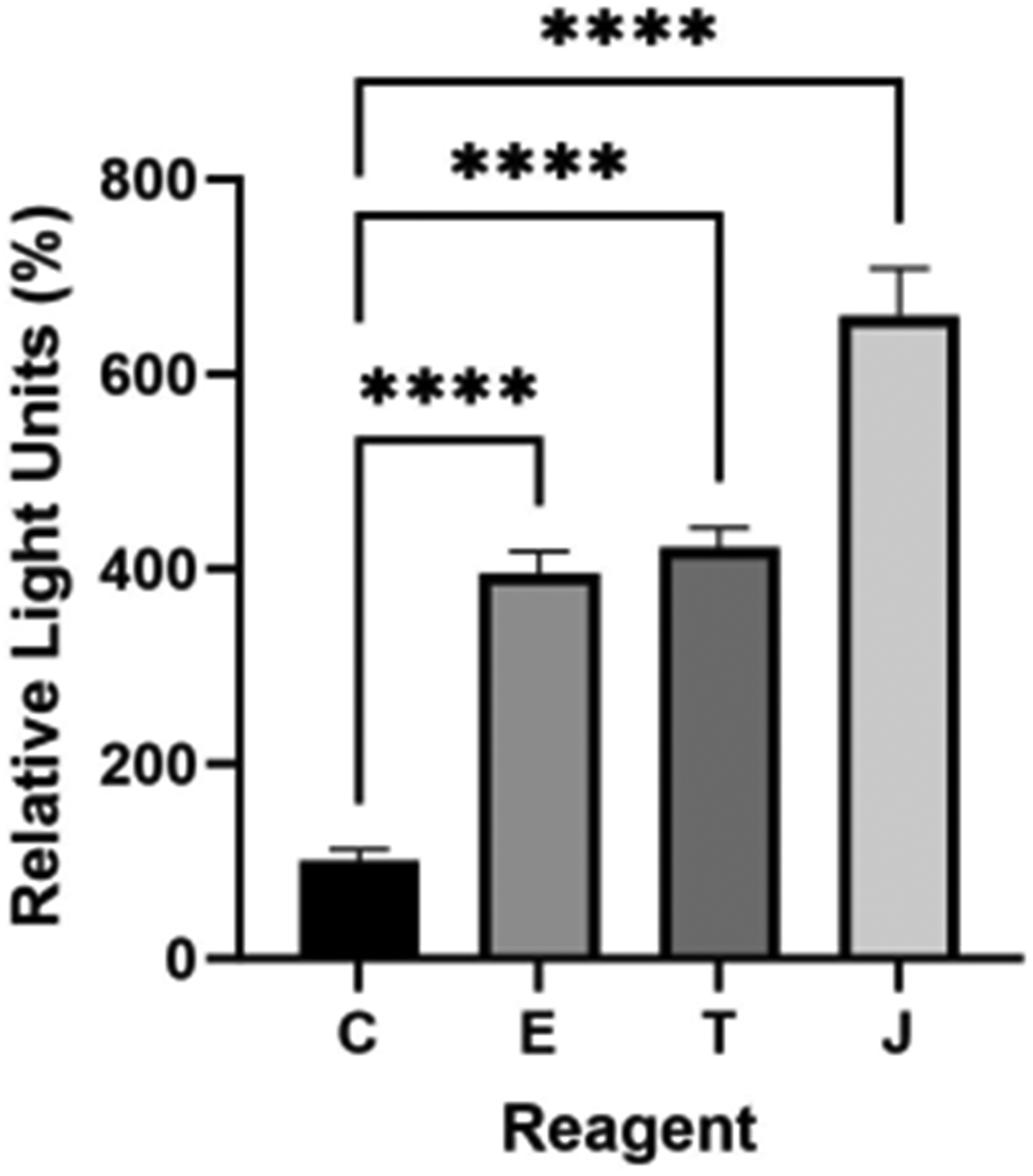
Luciferase expression 24 h after gWiz Luc delivery using proprietary reagents. C, control; E, Endofectin Max; T, Transit LT1, J, jetPrime. ****p < 0.0001 compared to control. n = 4 per group.
We next transfected differentiated C2C12 myotubes in situ with jetPrime. After 48 h, we examined the degree of transfection by detecting Katushka expression by fluorescence microscopy (Figs.3a and S1a). Cell death was not associated with transfection (Figs.3b and S1b). No significant transgene expression was observed in the naïve, pDNA only, and reagent only groups; the transfection efficiency in the DNA electrotransfer group was approximately 8% (Figs.3c and S1c).

Live and dead staining of differentiated myotubes transfected with pTurboFP635-C (Katushka) via jetPrime. (a) Fluorescence microscopy representative images of dead cells (green), live cells (blue), and transfected cells (red). Scale bar = 200 µm. (b) Quantification of the number of myotubes containing live (blue), dead (green), and red (transfected). (c) Quantification of transfected myotubes. n = 3–6 fields of view per group. ***p < 0.001, *p < 0.05.
pDNA delivery via electrotransfer
To examine whether C2C12 myotubes exhibited efficient pDNA transfection by electrotransfer, we electroporated differentiated C2C12 myotubes with pDNA (Figs.4a and S2a). Viability, as indicated by the number of myotubes containing blue nuclei, decreased by 35% (p < 0.0001) in the myotubes exposed to pDNA electrotransfer when compared to control myotubes (Figs.4b and S2b). No significant transgene expression was observed in the naïve, pDNA only, and pulse only groups; the transfection efficiency was approximately 36% in the group receiving pDNA electrotransfer (Figs.4c and S2c).

Live and dead staining of differentiated myotubes transfected with pTurboFP635-C (Katushka) via electroporation (EP). (a) Fluorescence microscopy representative images of dead cells (green), live cells (blue), and transfected cells (red). Scale bar = 200 µm. (b) Quantification of the number of myotubes containing live (blue), dead (green), and red (transfected). (c) Quantification of transfected myotubes. n = 3–6 fields of view per group. ***p < 0.001, *p < 0.05.
Chemokine expression
We quantified a subset of chemokine proteins secreted into the medium by differentiated myotubes 48 h after pDNA electrotransfer (Fig.5). After 8 days of differentiation, the myotubes were treated with pDNA and/or pulses. Protein expression in supernatants was evaluated using a multiplex bead array. Pulse application didnot induce chemokine expression. CXCL2 was downregulated after the introduction of pDNA. However, C-C motif chemokine ligand 3 (CCL3), CCL4, CCL5, and C-X-C motif chemokine ligand 10 (CXCL10) were significantly secreted after pDNA electrotransfer.
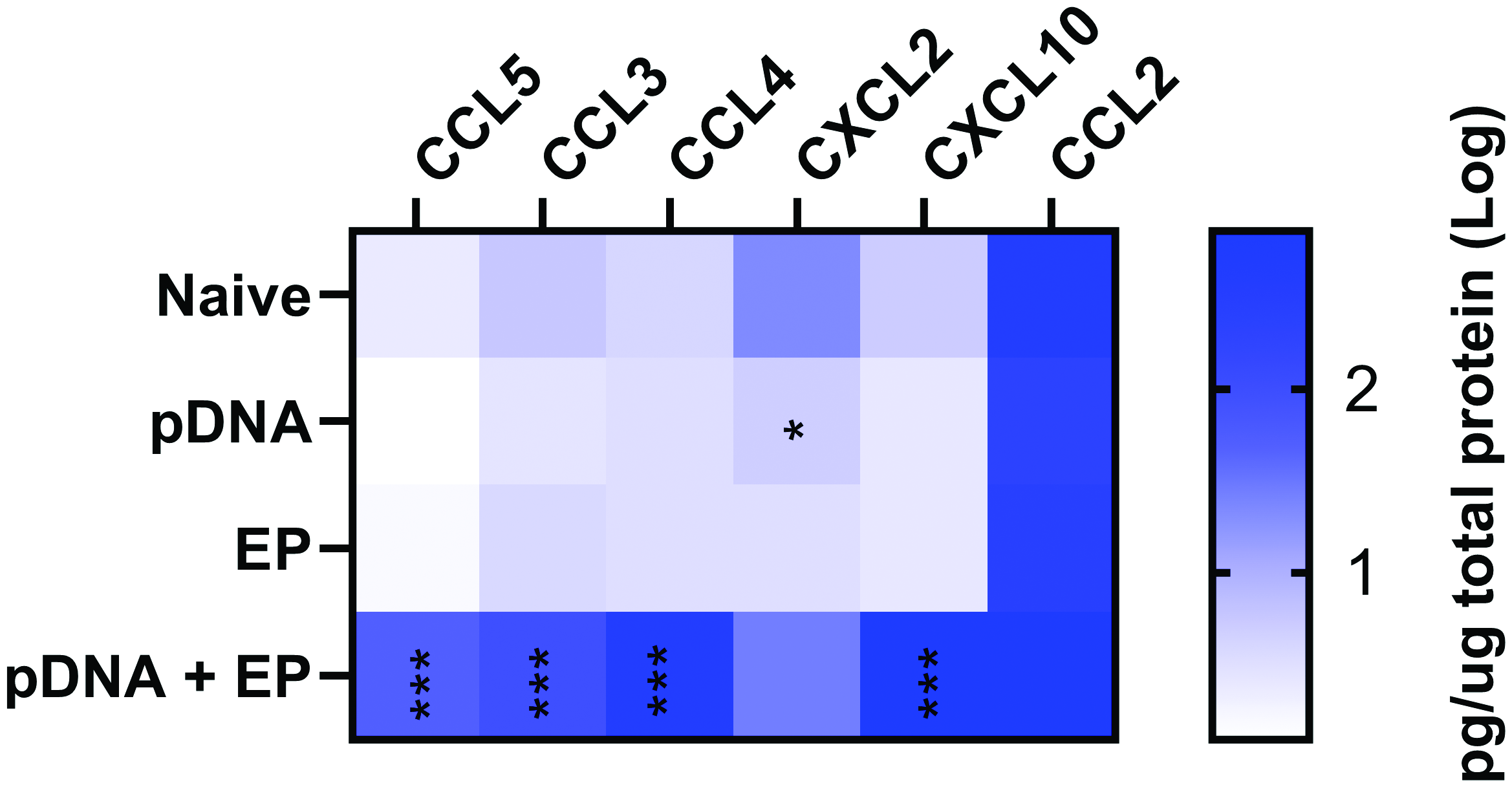
Cytokines and chemokines detected in mouse myotube medium 48 h after plasmid electroporation (EP). CCL, C-C motif chemokine ligand; CXCL, C-X-C motif chemokine ligand. ***p < 0.001, **p < 0.01, *p < 0.05 compared to control. n = 4–5 per group.
Discussion
Differentiation of murine C2C12 myoblasts is frequently used as a myotube model to mimic the invivo differentiation process invitro. C2C12 myotubes recreate many aspects of the development,2 contraction,3 and biomechanical properties4 of skeletal muscle myotubes, although direct comparisons are rare. With starvation, the majority of C2C12 cells pause in G1 after growth in differentiation medium, leave the cell cycle, and eventually differentiate and fuze to form multinucleated myotubes.13
Different proprietary formulations are complexed with DNA to enhance transfection. Transit LT1 (Mirus Bio) contains an amphipathic compound and a DNA-binding protein,18 EndoFectin Max (GeneCopoeia) is a lipid-based formulation, while jetPrime (PolyPlus Transfection) is a cationic polymer-based reagent. Each of these formulations successfully transfected C2C12 myotubes. Although high cell viability was maintained, the transfection efficiency was only 8%. Differentiated cells do not divide, and it is well-accepted that breakdown of the nuclear envelope during mitosis is optimal for nuclear entry of gene therapy vectors, particularly nonviral vectors.19,20 This may explain the level of transfection efficiency. Myotubes may transfect more easily during process associated with differentiation such as cell–cell membrane fusion. However, this study was designed to mimic fully differentiated rather than developing or regenerating muscle.
Myoblast plasmid transfection followed by differentiation into myotubes has been reported using electroporation,3,21,22 chemical methods,3,22–24 or viral methods.25 Sandri etal. reported the transfection of 3-day-old myotubes seeded on coverslips by electrotransfer.26 In this study, five 20 ms pulses at an applied voltage to distance ratio of 180 V/cmat 200 ms intervals were delivered using spatula-like electrodes. Luciferase expression and green fluorescence protein immunofluorescence staining were used to confirm transfection. Transfection efficiency was not measured. While our study varied in the pulse protocol, electrode, plasmid, and DNA concentration, our data confirmed the successful transfection of differentiated myotubes using pDNA electrotransfer. Myoblast transfection efficiency was previously published using two different pulse protocols and is approximately 20% as measure by flow cytometry.7 Interestingly, this efficiency may be lower than the observed myotube transfection level of 36%.
Plasmid DNA can be repeatedly electroporated intomuscles and cells due to its low immunogenicity,27 although acute inflammatory responses7,8 and inflammation9–11 have been reported. We observed the production of proinflammatory chemokines by myotubes. The expression pattern of myotubes did not necessarily mirror those of skeletal muscle 4 h after pDNA electrotransfer.8 Both myotubes and skeletal muscle secreted the chemokines CCL3, CCL4, and CXCL10 in response to pDNA electrotransfer. However, while myotubes secreted CCL5, skeletal muscle did not. Overall, skeletal muscle was much more responsive to pDNA exposure. Skeletal muscle produced increased CXCL2 in response to either pDNA injection or pDNA electrotransfer, while CXCL2 production was downregulated in myotubes with pDNA exposure. These differences may be due to the presence of other cell types invivo, the assay time point or the concept that tissue injection is different from simple DNA exposure in culture. Clearly, invitro systems may not entirely mimic tissue responses. The production of inflammatory chemokines generally supports the hypothesis that these responses may be due to the detection of plasmid DNA by endosomal or cytosolic DNA sensors such as cyclic guanosine monophosphate–adenosine monophosphate synthase and other putative sensors initiating inflammatory signaling.28,29 A multiplex bead array was used to detect expression of a therapeutic protein, IL-12p70, as well as its downstream effector interferon-γ, after intratumor electrotransfer.30
Finally, myotube stress may be induced by pulse application. By definition, pulse application induces permeabilized areas, commonly known as “pores,” in the cell membrane.31,32 Transcriptional changes in addition to inflammatory pathways are induced.33 The production of reactive oxygen species during pulse application has been described.34,35 It is well-established in several cell types that cells undergo transient cytoskeletal reorganization after pulse application.36–44
Conclusions
In many applications, myotubes differentiated invitro from myoblasts can be a potential model for skeletal muscle. Here, we demonstrate that myotubes can be successfully transfected in situ by electroporation. However, the biological responses of myotubes, as indicated by chemokine expression in this study, may not entirely parallel those of skeletal muscle. This may limit the translational importance of observations in this model.