
Abstract
Background:
The conformational p53-R175H is the most frequently occurring p53 mutant in cancers and a focus of mutant p53 targeted therapies. Plakoglobin is a dual cell adhesion and signaling protein with tumor suppressor activity. We previously showed that plakoglobin interacts with p53 and restores tumor suppressive activity of p53 mutants, including p53-R175H, in vitro.
Method:
Here, we used in silico modeling to predict the residues in p53-R175H that contribute the most to its interaction with plakoglobin, which identified p53-R175H Q167 and R248 as residues most involved in this interaction. To validate our in silico results, constructs were developed in which the predicted residues were substituted by alanine and transfected into the p53 null and plakoglobin deficient H1299 cells with or without plakoglobin. Transfectants expressing substituted residues were characterized using various biochemical and functional assays.
Results:
Co-immunoprecipitation and GST pull down assays showed a significant reduction in p53-R175H-plakoglobin association in p53-R175H-(Q167A, R248A and Q167A-R248A) transfectants. Despite the reduced interaction, in vitro invasion assays showed plakoglobin expression decreased invasiveness of all transfectants with the highest reduction in p53-R175H-R248A and R175H-Q167A-R248A expressing transfectants.
Conclusion:
These studies demonstrate, for the first time, the role of Q167 and R248 residues in p53-R175H's interaction with plakoglobin. The larger implication of these observations is the potential for exploring plakoglobin's interaction with p53-R175H mutant for the development of cancer therapeutics that restores the wild-type transcriptional activity/function of mutant p53s.
Introduction
The tumor suppressor and transcription factor p53 play a central role in the regulation of the cell cycle, apoptosis, DNA repair, senescence, metabolism, and immune system.1,2 In normal cells, p53 responds to cellular stress, such as DNA damage, hypoxia, and oncogenic insults, among other stimuli/insults, by regulating the expression of genes and pathways that maintain homeostasis and normal cellular functions.3–5 The inactivation or abnormal activation of p53 is one of the most frequent and effective strategies that cancer cells use to maintain survival and growth as well as the ability to invade normal tissues and metastasize.4 p53 is inactivated by several mechanisms including mutations, posttranslational modifications, and interactions with its primary endogenous inhibitor MDM2.6,7 p53 mutations are observed in >50% of all cancers and the majority of metastatic tumors.8 Consequently, restoring p53 activity is currently one of the most promising therapeutic strategies in fighting cancer.9
Majority of p53 mutations occur in the DNA-binding domain (DBD) of the protein with more than 80% being single missense mutations, 30% of which are represented by six hot spot mutations.10 The hot spot mutations are generally classified into DNA contact, which affect residues involved in the direct DNA–protein interaction [arginine (R), 273, 248] without conformational changes and structural/conformation mutations, which change the protein conformation [R (175, 245, 249, and 282)].10 Functionally, there are three types of p53 mutants: loss/partial loss of wild-type (p53-WT) function, dominant-negative (negatively regulates p53-WT), and gain of function (GOF; loss of tumor suppressor function and gain of oncogenic function).11,12 Strategies used for p53-targeted therapy are dependent on p53 status.9,13,14 For p53-WT loss of function mutants, therapies are generally based on drugs that target its interaction with its major negative regulator, MDM2. For GOF mutants, therapies are centered on drugs/small molecules that promote mutants p53 degradation, restore the wild-type transcriptional activity/function of mutants, and inhibit downstream oncogenic pathways activated by mutants.9,13,14
Plakoglobin (PG) is a dual adhesion and signaling protein that we have identified as a novel interacting partner of p53.15–18 Previously, we have shown that PG interacted with p53-WT and several p53 mutants, including p53-R175H.15–18 p53-R175H is the most frequently occurring p53 mutant in various cancers, with potent GOF oncogenic properties.19 Coexpression of PG in cells expressing p53-R175H restored its tumor suppressor function invitro by significantly reducing cell proliferation, migration, and invasive properties.17 To gain further insights into p53-R175H-PG interaction and its potential therapeutic application, we used in silico 3D molecular dynamic modeling, which predicted Q167 and R248 residues critical in mediating p53-R175H interaction with PG. We then performed invitro analyses using site directed mutagenesis to test the validity of the predicted model.
Our results suggested that Q167 and R248 amino acid residues are important for p53-R175H interaction with PG and their respective substitution with alanine individually or together reduced p53-R175H-PG interaction. Interestingly, despite the reduced interaction of PG with p53-R175H, PG coexpression with different p53-R175H mutated constructs decreased their invasiveness to various extent. Overall, the results showed that Q167 and R248 residues in p53-R175H are important for its interaction with PG. These results support the consequences of PG association with mutant p53 and provide some insights that will be beneficial for the development of potential cancer therapeutics that could function by mimicking PG effects in cancers expressing this type of mutant p53.
Materials and Methods
In Silico Modeling of p53-R175H and PG Interaction
Cothreading of p53-R175H and PG
COTH webserver20 was initially used to predict the complex structure of p53-R175H core domain and the C-terminal residues of PG (Fig.1A). All 10 predictions from COTH were of the p53 core-domain in a tetramer assembly with protein data bank (PDB) IDs: 3KMD, 3D0A, 3EXJ, 4IBU, 1TSR, 3EXL, 4GUO, 1TUP, 4G82, and 1KZY. Since the COTH webserver only outputs the alpha-carbon coordinates of the query residues, the top prediction based on PDB ID: 3KMD21 was used as a starting point to build the model. More specifically, COTH predictions yielded the alpha-carbon coordinates of p53-R175H core domain and residues 724-736 of PG. Tleap of Ambertools1822 was then used to place the missing atoms for each residue to build an all-atom model from the predicted alpha-carbon positions.

(A) Schematic representation of p53 and plakoglobin structures. DNA binding domain (DBD) of p53 interacts with C-terminal domain of plakoglobin (delineated by red dashed lines). (B) Plot demonstrating the number of conformations of the p53-R175H-plakoglobin complex in each cluster and the corresponding average binding energies of each cluster. Cluster 6 (red arrow) has the lowest binding energy of −49 kcal/mol. MMGBSA: mechanics-generalized born surface area.
Construction of the p53-R175H-PG Complex Structure
There is a single experimentally resolved structure of PG of residues 126-673 (PDB ID: 3IFQ).23 It was necessary to construct the missing C-terminal residues of PG involved in the interaction with p53. Therefore, Quark webserver24,25 was used for the ab initio structure prediction of residues 674-723 and 737-745 of PG, which were not mapped by COTH. A model of PG residues 126-745 was then constructed by joining residues 126-673 from PDB ID: 3IFQ.23 Quark-predicted residues 674-723 followed by COTH-predicted residues 724-736 in their relative positions with respect to p53-R175H. Quark-predicted residues 737-745 were then joined to residue 736.
p53-R175H-PG complex was then solvated in a neutralized 12 Å TIP3P water box with 0.15 M NaCl and parameterized using Amberff114SB force-field. To increase conformational sampling of the complex, we used accelerated molecular dynamics (aMD) using Amber software.26 For this, the solvated complex was first minimized and then gradually heated from 0 to 310 K using heavy restraints on the backbone atoms. Restraints were gradually decreased until they were completely removed before the complex was simulated for 150 ns using classical molecular dynamics (cMD) to obtain parameters required for aMD. The latter simulation was run for 200 ns.
Trajectory Processing
Root-mean-square-deviation (RMSD) based clustering of 10,000 frames obtained from aMD was clustered based on a 2 Å cutoff for residues 670-745 of PG using CPPTRAJ of Ambertools18.22 This yielded 101 different clusters that represent distinct complex conformations sampled during the simulation. The average Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) binding energy of each cluster was calculated using MMPBSA.py.27 Additionally, the pairwise binding energy decomposition was also calculated.
Cell Lines and Culture Conditions
All tissue culture reagents were purchased from Gibco unless stated otherwise. H1299, the non-small cell lung carcinoma cell line deficient in p53 and PG expression, was provided by Dr. Roger Leng, University of Alberta, and has been described previously.15,17,28 H1299 cells and its transfectants expressing PG and various p53-R175H constructs were maintained in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (HyClone Laboratories, USA), 1% penicillin-streptomycin, and 5 µg/mL kanamycin (complete MEM, CMEM).
Plasmid Construction and Transfection
Mutant p53 constructs were generated from HA-tagged p53 cDNA as described previously.15,17 Site-directed mutagenesis was used to generate plasmids encoding p53-R175H and p53-R175H in which glutamine (Q) 167 or arginine (R) 248 or both were mutated to alanine to express mutants p53-R175H-Q167A, p53-R175H-R248A, and p53-R175H-Q167A-R248A (Synthego Corporation, USA). The authenticity of all constructs was validated by sequencing both at Synthego and the University of Alberta Molecular Biology Service Unit.
Transfectants were generated using the jetOPTIMUS® (Polyplus, France) DNA transfection reagent following the manufacturer’s protocol. Cells were transfected with 2 µg of plasmids encoding various p53-R175H constructs alone or together with 3 µg of PG. For transient transfections, cultures were processed within 48 hours for various assays. Stable transfectants were selected by supplementing CMEM with 800 µg/mL G418 (p53 transfectants) or 800 µg/mL G418 and 600 µg/mL hygromycin (PG-p53 transfectants) 48 hours post-transfection. Multiple single cell clones for each transfectants were obtained using limiting dilution, and the expression of p53 or coexpression of p53 and PG was verified using immunofluorescence and immunoblot assays. All experiments were repeated at least three times and conducted with stable transfectants, unless indicated otherwise.
Preparation of Total Cell Extracts and Immunoblotting
Confluent 100 mmculture dishes were rinsed with cold PBS and extracted with 1 mL RIPA lysis buffer [150 mM NaCl, 50 mM Tris-HCl pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA, 1 mM PMSF, 1 mM NaF, 1 mM Na3VO4, and Roche protease inhibitor cocktail (Sigma, Canada)] for 20 minutes on a rocker at 4°C. Cells were scraped and centrifuged at 48,000×g for 10 minutes, and supernatants were processed for immunoblotting. Equal amounts of total cellular lysates (100 µL) were solubilized in 20 µL hot 6X-SDS sample buffer [10 mM Tris-HCl pH 6.8, 2% (w/v) SDS, 50 mM dithiothreitol (DTT), 2 mM EDTA, and 0.5 mM PMSF], boiled for 10 minutes. Proteins were resolved by SDS-PAGE, transferred onto nitrocellulose membranes, and processed for immunoblotting with primary and secondary antibodies at concentrations indicated in Table1. Membranes were scanned using an Odyssey CLx infrared imaging system. Comparable loadings among various cell lines were confirmed by immunoblotting of the same membrane with anti-actin antibodies.
Antibodies and their dilutions in various assays
Immunofluorescence
Cells were grown to 80% confluency on glass coverslips and processed for immunofluorescence staining as described previously.15,18 Briefly, coverslips were rinsed with cold PBS, fixed with 3.7% formaldehyde solution, and permeabilized with cytoskeletal extraction buffer (50 mM NaCl, 300 mM sucrose, 10 mM PIPES pH 6.8, 3 mM MgCl2, 0.5% Triton X-100, 1.2 mM PMSF, and 0.1 mg/mL DNase and RNase) for 7 minutes. Coverslips were blocked with 4% goat serum and 50 mM NH4Cl in PBS, rinsed with PBS-BSA, and incubated with primary antibodies for 1 hour, followed by 30 minutes in species-specific secondary antibodies (Table1). Nuclei were stained with 1:2000 DAPI. Coverslips mounted in elvanol containing 0.2% paraphenylene diamine (PDD, w/v) and viewed using a 40× objective of a Zeiss fluorescent microscope.
Coimmunoprecipitation
All steps were carried out at 4°C. Confluent 100 mmculture dishes were extracted with 1 mL RIPA buffer as described earlier. Cells were removed from the plates and centrifuged at 48,000×g for 10 minutes. The resulting supernatant was divided into equal aliquots. Duplicate aliquots were incubated with 50 µL protein A/G (Pierce, Canada), antip53, or anti-PG antibodies (Table1) and incubated overnight on a rocker-rotator. Beads containing immune complexes were washed threetimes with RIPA buffer and immune complexes eluted in hot 4X-SDS sample buffer. Equivalent amounts of total cellular proteins immunoprecipitated from eachcell line were loaded onto SDS-polyacrylamide gels and processed for immunoblot using p53 and PG antibodies.
Preparation of PG-GST
A construct encoding pGEX-TEV-PG was kindly provided by Dr. William Weis.23 To express PG-GST protein, Escherichia coli DH5α cells were transformed by pGEX-TEV-PG constructs. Transformed cells were grown in Luria-Bertani broth at 37°C to an A600 of 0.6–0.8 and induced with 0.5 mM IPTG (isopropyl-β-d-1-thiogalactopyranoside, Thermo Fisher, Canada). Cultures were grown for an additional 6 hours at 30°C, harvested by centrifugation at 4,000 × g at 4°C for 12 min, and the supernatants discarded. Pellets were resuspended in 6 mL of cold bacterial lysis buffer (500 mM NaCl, 0.5% NP-40, 50 mM Tris-HCl pH 7.6, 5 mM EDTA, 5 mM EGTA, 1 mg/mL lysozyme, 10 mM DTT, 2.5 U/mL DNase, 1 mM PMSF, and an EDTA-free protease inhibitor cocktail tablet) and lysed via sonication. Lysates were centrifuged at 10,000 × g for 25 min at 4°C, and the supernatants were divided into 1 mL aliquots, snap frozen, and stored at −80°C.
PG-GST Purification and GST Pull-Down Assay
A 1 mL aliquot of PG-GST bacterial lysate was incubated with 150 µL of glutathione sepharose beads (GE Life Sciences) on a rocker-rotator at 4°C for 6 hours. Beads were centrifuged at 21,000 × g for 1 minutes, and supernatants were aspirated. Beads were washed 3× using cold KCl-PBS (0.137 M NaCl, 0.0027 M KCl, 0.01 M Na2HPO4, and 0.0018 M KH2PO4) and stored in KCl-PBS at 4°C.
Cells from 100 mmcultures of H1299 and H1299-p53-(R175H, R175H-Q167A, R175H-R248A, and R175H-Q167A-R248A) transfectants were extracted in 1 mL RIPA lysis buffer for 20 min on a rocker-rotator at 4°C. The lysates were centrifuged at 48,000 × g for 10 min at 4°C, and supernatants were removed. For pull down assays, 900 µL of cell lysates was mixed with 40 µL PG-GST or GST (control) beads and incubated on a rocker rotator for 4–6 hours at 4°C. The beads were washed 3× with PBS-KCl buffer (0.137 M NaCl, 0.0027 M KCl, 0.01 M Na2HPO4, and 0.0018 M KH2PO4), eluted with 4× SDS sample buffer, and processed for immunoblot using p53 and PG antibodies.
In vitro Invasion Assays
Matrigel invasion assays were performed according to the manufacturer's protocol (Corning, Life Sciences). For each cell line, 5 × 104 cells in 0.2 mL serum-free media were plated in the top compartment of Matrigel-coated invasion chambers (8 µm pore PET membrane). Fibroblast conditioned media (0.8 mL) was added to thebottom chambers, and plates were incubated overnight at 37°C in 5% CO2. After 24 hours, inserts werestained using NovaUltraTM Hema-Diff Stain Kit (IHC World Life Science Products & Services, USA), following manufacturer’s protocol. Following staining, inserts were viewed under an inverted microscope using a 20× objective lens and photographed. The number of invaded cells was counted in seven random fields for each membrane using the NIH ImageJ Cell Counter software. The results are presented as means ± SD in histograms.
Statistical Data Analysis
All quantitative data were presented as mean ± standard deviation (SD). Statistical significance between groups was assessed using Student’s t-test for all assays. All biochemical experiments were repeated at least 3-6 times, and the figures are representative of one typical experiment for each assay. All functional assays were repeated at least three times, and the histograms represent the average of all assays.
Results and Discussion
Previously, we showed that PG interacts with wild type and mutant p53 via the DNA binding domain (DBD) of p53 and C-terminal domain of PG.15 We also demonstrated that PG’s interaction with several mutant p53forms including p53-R175H, the most frequently occurring p53 mutant, restored their tumor suppressor activity invitro.15–18,29–31 p53-R175H is a structural/conformational mutant that changes the DBD conformation of wild type p53, which not only disrupts its interaction with target DNA thereby attenuating its tumor suppressor activity but also bestows p53-R175H with potent GOF activity.10–12,32 Notably, when coexpressed, PG attenuates the oncogenic functions of p53-R175H as demonstrated by the significantly reduced growth, migratory, and invasive properties of p53-R175H expressing cells in the presence of PG, which was also associated with suppression of oncogenic genes expression such as c-MYC and S100A4.17 As such, we hypothesized that PG interaction with conformational mutants like p53-R175H conceivably changes their conformation and restores their binding to the promoters of wild type p53 target genes. In this study, we used in silico modeling to identify potential amino acid residues that mediate p53-R175H interaction with PG, and molecular techniques to validate this interaction.
In Silico Modeling Identified p53-R175H Q167 and R248 as Residues Critical for its Interaction with PG
Previously, we had shown that the DBD of p53 mediated its interaction with the C-terminal domain (674-745) of PG.15 In this study, we sought to identify the residues via which the conformational p53-R175H mutant binds to PG. Since there are no experimentally resolved PG C-terminal structures, we utilized the COTH webserver to predict the relative position of p53-R175H DBD to PG. This yielded the alpha-carbon positions of residues 92-291 of p53-R175H and residues 724-736 of PG. Quark ab initio webserver was then used to predict the structure of residues 674-723 and 737-745 that constitute the missing residues of PG C-terminal domain. To construct the protein complex, we assembled the experimentally resolved structure of armadillo domains—residues 126-673 of PG (PBD; ID: 3IFQ), the Quark ab initio (Quark. Bio.tools) predicted structures of residues 674-723 and 737-745, and the COTH-predicted residues of 724-736.23
The initial structure of the complex was built by adding the missing residue atoms to the predicted alpha carbons. This initial atom placement was done using tleap resulting in a structure with a lot of steric clashes and unequilibrated bond lengths and angles. Due to the crudeness of the initial structure and the intrinsically disordered nature of the C-terminal domain of PG, it was important to equilibrate the protein structures and efficiently sample the conformational landscape of the complex. We, therefore, ran cMD for 150 ns for equilibration and to obtain parameters for aMD, which was performed for 200 ns. Sampled conformations were clustered based on the C-terminal residues closest to p53 with a RMSD cutoff of 2 Å to distinguish distinct complex conformations. A total of 101 clusters were obtained. The MM-GBSA binding energy of each cluster was calculated. The distribution of the size of each cluster and its average binding energy is shown in Fig.1B. Cluster 6 had the lowest average MM-GBSA binding energy between p53-R175H and PG of −49 kcal/mol (Fig.1B, red arrow). We also decomposed the calculated average binding energy to identify the residue pairs that are most involved in the p53-R175H-PG interaction (Table2).
Lowest molecular mechanics-generalized born surface area (MMGBSA) binding energy (kcal/mol) of the residue pairs in the modeled p53-R175H-plakoglobin complex
The representative structure of cluster 6 is shown in Fig.2A. Several hydrogen bond networks can be observed at the interaction interface of the two proteins (Figs.2B and C). Specifically, hydrogen bonds are formed between the side-chains of Q167 (p53) and D719 (PG), while the backbone of D719 forms an intrachain hydrogen bond with the side-chain of H717 (Fig.2B). Additionally, a hydrogen-pi interaction is formed between the side-chain of H717 (PG) and backbone of L93 of p53 (Fig.2B). Furthermore, a dense hydrogen-bond network between residues P692 (PG)- R174 (p53)-E171 (p53)- N690 (PG)-G245 (p53)-N247 (p53) is formed, which stabilizes the interaction between the two proteins. Additionally, we observed a salt bridge formed between R248 of p53 and E654 of PG and another between R174 and E171 of p53 (Fig.2C). Based on the contributions of the individual residues to the total MMGBSA binding energy (Table2), R248 and Q167 of p53-R175H contributed the most to the total binding energy of the complex.
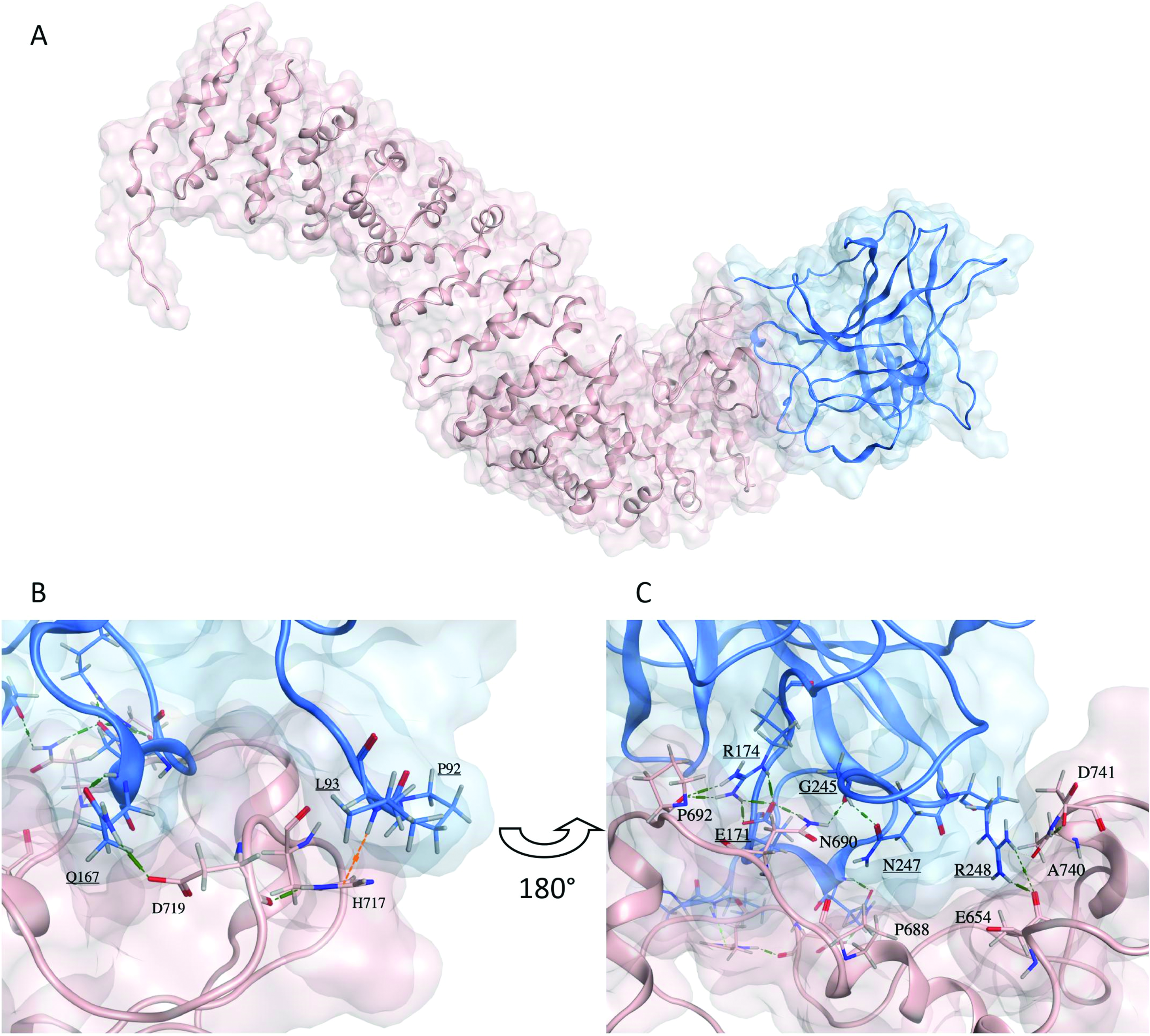
(A) Predicted representative structure of the cluster with the lowest calculated MMGBSA of plakoglobin (pink) in complex with p53-R175H (blue). (B and C) Stick representation of the interaction interface residues of plakoglobin (pink sticks) and p53-R175H (blue sticks and underlined labels). Hydrogen bonds are shown in green dashed lines, and a hydrogen-pi interaction is demonstrated by an orange dashed line.
To validate this prediction, site-directed mutagenesis was performed to substitute Q167 and R248 residues individually or together by alanine (A). H1299 cells expressing the p53-R175H-Q167A and R248A mutated forms alone or together (p53-R175H-Q167A-R248A) with PG were then examined for p53 and PG expression and localization patterns as well as their interaction. We also assessed for the effect of PG expression on the oncogenic functions of the mutated p53-R175H-Q167A, R248A, and R175H-Q167A-R248A forms.
Expression and Subcellular Localization of p53-(R175H, R175H-Q167A, R175H-R248A, and R175H-Q167A-R248A) with and without PG in H1299 cells
Plasmids encoding p53-R175H-Q167A, p53-R175H-R248A, and p53-R175H-Q167A-R248A were used to develop transient and stable transfectants expressing various forms of p53-R175H alone or together with PG in H1299 cell line. This cell line is uniquely suited for these analyses since it does not express endogenous PG and p53.
Western blot analyses (Fig.3A) using p53 and PG antibodies and total cellular extracts (TCEs) from transfectants expressing p53-R175H mutants alone or together with PG confirmed similar expression of both proteins in various transfectants. Immunofluorescence staining using p53 antibodies showed primarily nuclear localization of p53 in cells expressing various p53-R175H constructs only (Fig.3B, PG). Double immunofluorescence staining with p53 and PG antibodies in transfectants coexpressing both proteins (Fig.3B, +PG) showed that while p53 was mainly localized to the nuclei, it was also detected in the cytoplasm. This p53 cytoplasmic staining was more prominent in H1299-PG-p53-R175H and PG-p53-R175H-R248A cells relative to H1299-PG-p53-R175H-Q167A and PG-p53-R175H-Q167A-R248A transfectants (Fig.3B, +PG).
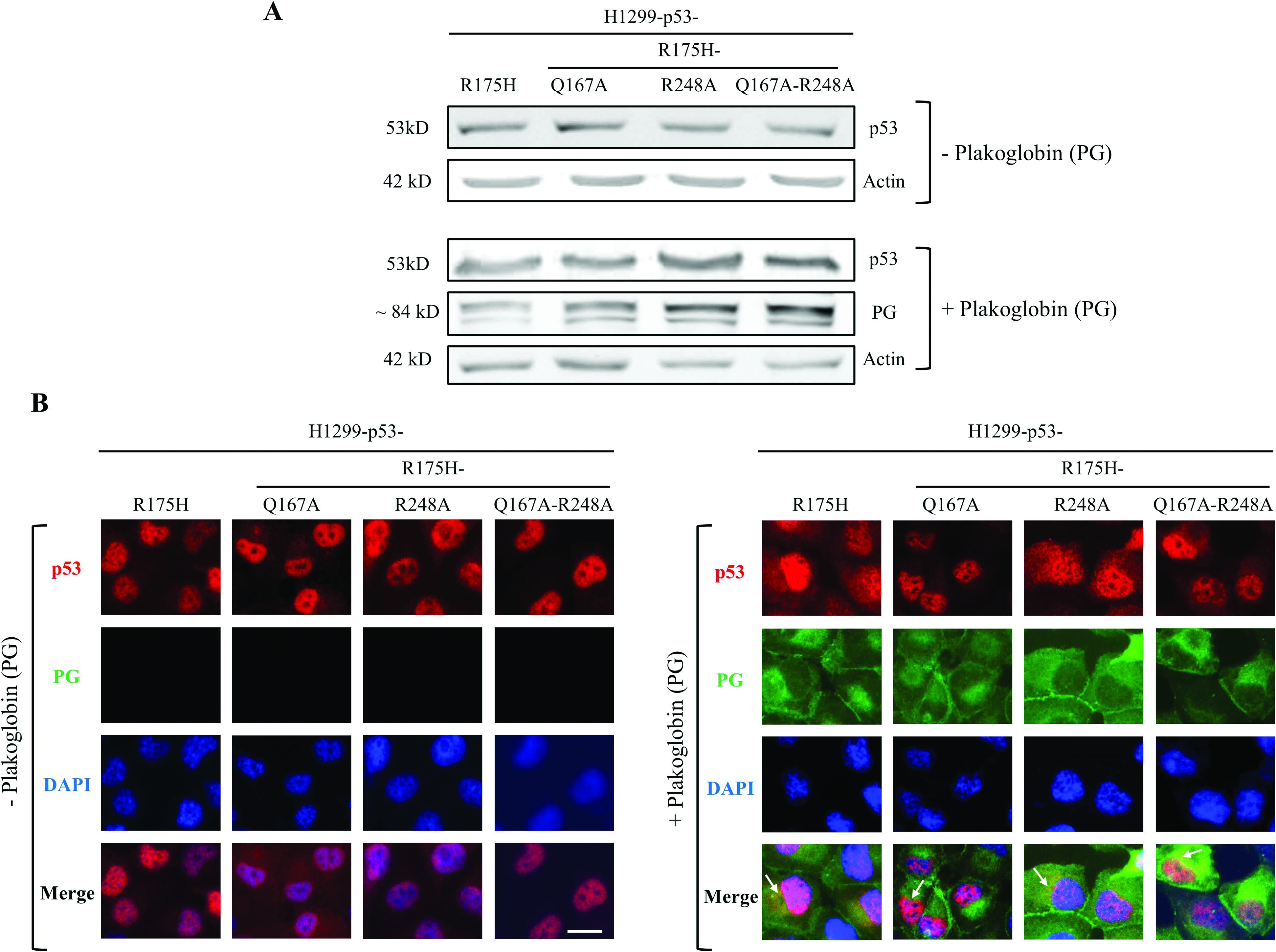
p53 and plakoglobin (PG) protein expression and localization in H1299-p53-(R175H, R175H-Q167A, R175H-R248A, and R175H-Q167A-R248A) transfectants. (A) Total cellular extracts from H1299 transfectants expressing various forms of p53-R175H without [− plakoglobin (PG)] or with [+PG] were processed for immunoblot using p53 and plakoglobin antibodies. Beta-actin was used as a loading control. (B) Stable H1299 transfectants expressing various forms of p53-R175H without [−PG] or with plakoglobin [+PG] were processed for immunofluorescence staining using p53 (red) and plakoglobin (green) primary antibodies followed by species-specific secondary antibodies. Nuclei were stained with DAPI (blue). Arrows indicate colocalization of plakoglobin and p53-R175H in H1299-PG-p53-R175H cells. Bar, 7 µm.
PG staining in all H1299-PG-p53-R175H transfectants showed distinct membrane localization and cytoplasmic distribution typical of this protein. The membrane localization is consistent with PG’s role incell–cell adhesion via its participation in the formation of adherens junction and desmosomes, whereas itscytoplasmic/nuclear distribution is associated withits signaling functions.33 There was distinct codistribution of PG and p53 in all transfectants (Fig.3B, +PG, arrows), which is consistent with our previousstudies that demonstrated colocalization of p53 and PG.15–17 Together, these results showed that substituting Q167 and R248 individually or together by alaninein p53-R175H does not interfere with thep53-R175H protein expression and subcellular localization.
Substitution of Q167 and R248 Residues by Alanine Protein Reduced p53-R175H Interaction with PG
We then performed coimmunoprecipitation (Fig.4A) and GST pull down (Fig.4B) experiments to examine whether substituting Q167 and R248 with alanine individually or together in p53-R175H affected its interaction with PG. H1299 cells were transiently cotransfected with PG and the various p53-R175H constructs. Twenty-four to 48 hours post-transfection, an equal amount of TCE from the respective transfectants was processed for sequential immunoprecipitation and immunoblotting with p53 and PG antibodies. Immunoblotting of TCEs from the various p53-R175H transfectants showed similar levels of p53 and PG expression among all PG-p53-R175H transfectants [H1299-PG-p53-(R175H, R175H-Q167A, R175H-R248A, and R175H-Q167A-R248A)] (Fig.4A, TCE, IB: p53, PG, actin).

Substitution of Q167 and R248 residues by alanine (A) in p53-R175H reduced its interaction with plakoglobin (PG). (A) Total cellular extracts (TCE) from H1299 cells coexpressing plakoglobin with various forms of p53-R175H, R175H-(Q167A, R248A, Q167A, and R248A) were processed for sequential immunoprecipitation (IP) and immunoblotting (IB) using p53 and plakoglobin antibodies. (B) TCEs from H1299 cells expressing various forms of p53-R175H were incubated with GST-tagged plakoglobin (PG-GST) and processed for GST-pull down assays (PG-GST PD) and immunoblotting (IB) with p53 and plakoglobin antibodies. Relative intensity of p53 protein bands coimmunoprecipitated with plakoglobin or pulled down with PG-GST was quantitated using NIH ImageJ program. Beta-actin was used as a loading control.
Immunoprecipitation assays detected comparable levels of p53 in various transfectants when p53 immunoprecipitates were blotted with p53 antibodies. In contrast, immunoblotting of PG immunoprecipitates with p53 antibodies detected significantly lower levels of p53 coprecipitated with PG in p53-R175H-(Q167A, R248A, and Q167A-R248A) expressing cells relative to PG-p53-R175H transfectants. Compared to p53-R175H, the PG coprecipitated with p53-R175H-Q167A, p53-R175H-R248A, and p53-R175H-Q167A-R248A was reduced by 70%, 60%, and 80%, respectively (Fig.4A, IP).
Results of coimmunoprecipitation studies were further confirmed by GST-pull down assays (Fig.4B). Here, we used PG-GST beads to pull down p53 from equal amounts of cellular lysates of transfectants expressing p53-R175H and p53-R175H-(Q167A, R248A, and Q167A-R248A). As shown in Fig.4B, relative to p53-R175H, the amount of p53 pulled down by PG-GST beads was reduced by 30%, 50%, and 60% in p53-R175H-Q167A, p53-R175H-R248A, and p53-R175H-Q167A-R248A transfectants, respectively. Overall, the results of these two independent assays showed that Q167 and R248 substitution by alanine reduced the binding of PG to p53-R175H. Depending on the assay type, cells expressing p53-R175H-Q167A or R248A or both showed reduced interaction with PG by a minimum of 30% and maximum of 80%. These results also demonstrated that neither single nor double mutation was able to completely abolish PG-p53 interaction.
PG coexpression Decreased Invasiveness of p53-R175H-(Q167, R248, and Q167-R248) Expressing H1299 Cells
We have previously demonstrated that PG interaction with several p53 mutants in various carcinoma cell lines restored mutant p53 tumor suppressor function invitro. Specifically, we have shown that p53-R175H expression in H1299 cells increased cellular migration and invasion, while PG coexpression significantly reduced these properties by >70%.17 Based on these observations and since invasion is the ultimate step in the metastatic cascade, preceded by motility and migration, we performed invasion assays to determine if the reduced PG interaction with p53-R175H-(Q167A, R248A, and Q167A-R248A) affected the invitro invasiveness of these transfectants.
In the absence of PG, there was no difference in the invasiveness of H1299-R175H-Q167A and H1299-R175H transfectants. In contrast, the number of invaded cells was increased by ∼43% in both H1299-p53-R175H-R248A and p53-R175H-Q167A-R248A compared to H1299-R175H transfectants (Fig.5). The increased invasiveness of p53-R175H transfectants expressing R248A substitution may be explained by the essential role of R248 residue in mediating direct binding of p53-WT to DNA. Substitutions in R248 are contact hotspot mutations, and many of them have been shown to be GOF and activate tumorigenic and metastatic pathways.10,34–36 Among various mutations in R248 residue (R248W, R248Q, R248P, R248G, and R248L), p53-R248L, in which arginine is mutated to leucine (L), is biochemically similar to alanine substitution used in the current study, and R248L mutant has been shown to possess increased oncogenic functions.37,38 It is therefore conceivable that the increased invasiveness observed in p53-R175H-R248A and p53-R175H-Q167-R248A transfectants may be due to a probable acquisition of new oncogenic properties as a result of the R248A mutation, which may be caused by the presence of a conformational and a contact mutation in the same protein.

Plakoglobin (PG) coexpression decreased invitro invasiveness of H1299-p53-R175H and H1299-p53-R175H-(Q167A, R248A, and Q167A-R248A) transfectants. Twenty-four hour invasion assays were performed in triplicate in Matrigel-coated transwell membranes. Membranes were fixed and stained, and the number of invaded cells was counted in seven random fields for each membrane using the ImageJ Cell Counter program. Values for H1299-(p53-WT, PG, and p53-PG) transfectants were normalized to H1299 cells, whereas values for H1299-(p53-R175H and PG-p53-R175) transfectants were normalized to H1299-p53-R175H cells and averaged. Histograms represent the normalized average
When PG was coexpressed, invasiveness of H1299-PG-(p53-R175H and -p53-R175H-Q167A) was significantly reduced by ∼80%. There was also a greater drop in the cellular invasiveness of H1299-PG-(p53-R175H-R248A and p53-R175H-Q167A-R248A) expressing cells (∼120%), indicating that PG coexpression not only reduced invasiveness of p53-R175H or p53-R175H/Q167A expressing cells but also nullified the intensified oncogenic effects induced by the R248A substitution. In addition to reducing the oncogenic functions of mutant p53, we have shown that PG promotes the tumor suppressive functions of p53-WT,15 which is also demonstrated herein (Fig.5). Notably, individual expression of p53-WT or PG in H1299 cells decreased invasiveness by ∼35% and 20%, respectively,15 indicating that PG displays some tumor suppressive activity independent of p53-WT. This tumor suppressive role of PG is greatly enhanced in the presence of p53-WT as revealed by the >70% decreased invasiveness of H1299 cells upon coexpression of PG and p53-WT (Fig.5 and Ref. 15). These observations suggested that (1) PG had p53-independent and dependent tumor suppressive activity, (2) its tumor suppression effect was synergistic with that of p53-WT expression, and (3) PG coexpression not only reduced invasiveness of p53-R175H or p53-R175H/Q167A expressing cells but also nullified the intensified oncogenic effects induced by the R248A substitution.
In summary, our data from in silico modeling and the experimental results indicate that amino acid residues Q167 and, more significantly, R248 are important for p53-R175H interaction with PG. These substitutions decreased but did not completely inhibit p53-R175H-PG interaction. Invasion assays revealed that cells expressing p53-(R175H and R175H/Q167A) exhibited similar invasiveness, whereas transfectants expressing p53-(R175H/R248A or R175H/Q167A/R248A) showed increased invasiveness relative to p53-R175H cells. PG coexpression reduced invasiveness of all p53-R175H alanine mutated transfectants, and this can be explained by the potential fractions of p53-PG complexes still present in these transfectants (Fig.4B) that may still be able to carry out tumor suppressive functions. Overall, our results indicate the importance of the amino acid residues identified in p53-R175H that mediate its interaction with PG.
Conclusion
Currently, a considerable effort is being invested to develop p53 activating drugs since the majority of tumors express GOF or inactive mutant p53 protein.39–42 One such therapeutic strategy focuses on developing drugs/small molecules that restore the wild-type transcriptional activity/function of mutant p53s and inhibit their respective activated downstream oncogenic pathways.43–50 PG interaction with several p53-mutants has been shown to attenuate their oncogenic functions and restore their tumor suppressive roles. The studies described here shed light on the residues in one of the most frequent GOF p53 mutant (p53-R175H) that is involved in its interaction with PG. This work provides evidence for exploiting PG-p53 interaction for potential targeted therapeutic purposes. The advantage of developing drugs that mimic the effects of PG on mutant p53 to restore their tumor suppressor activity is that these compounds may have the benefit of representing a naturally occurring biological molecule that positively regulates p53 tumor/metastasis suppressor function with fewer potential side effects.
Footnotes
Acknowledgments
The authors would like to thank Dr. William Weis for the PG-GST construct and Dr. Rashmi Panigrahi for her critical review of the manuscript.