
Abstract
Background:
Cytochrome P450 11β-hydroxylase (H11b) converts testosterone (T) into 11-hydroxytestosterone. In Clarias batrachus (catfish), H11b occurs in regular form and has three variants as well. The variant forms contain the complete steroid binding site, though they lack other regions. Thus, they are suggested to be involved in regulating steroid concentration.
Method:
The structure of H11b and its three variants, Var1, Var2 and Var3 were modeled by homology modeling using human cholesterol side-chain cleavage enzyme, CYP11A1 as a template. The binding of T with the H11b in holo and apo (non-heme state) forms and with all three variants was studied using docking. Furthermore, molecular dynamics simulations of the protein-ligand complexes were carried out and the binding energies of T were calculated using the molecular mechanics-Poisson–Boltzmann surface area (MM-PBSA) method.
Results:
The analysis showed that the regular protein in its apo-form had the largest binding affinity with T followed by holo-form and Var1. The higher binding affinity was found to be driven by van der Waals interactions. Var3 had the least binding affinity possibly due to the high polar solvation energy.
Conclusion:
The differences in the binding affinities suggest that the competitive binding of T between H11b and its variants could be the key factor in regulating the synthesis of 11-hydroxytestosterone.
Introduction
Cytochrome P450 11β-hydroxylase (H11b) is a steroid enzyme involved in the hydroxylation of testosterone (T) into 11-hydroxytestosterone (OHT). OHT is a precursor for the biosynthesis of 11-ketotestosterone (11-KT), a potent fish androgen. 11-KT and T are the two major androgens that regulate various processes including reproduction in teleost.1 Biosynthesis of 11-KT occurs in testicular Leydig cells in addition to its peripheral conversion in the adrenal and liver.2 11-KT induces male sexual phenotype, secondary sexual characteristics, and female-to-male sex reversal.3 It is also involved in testicular growth, spermiation, and recrudescence in fish.4
Teleost (Teleostei), one of the subdivisions of the class actinopterygii (bony fishes), belongs to a large and extremely diverse group of ray-finned fishes. Teleost fishes constitute the largest group of vertebrates and are most widely used models for studies on endocrinology, physiology, and evolution. Catfishes, one of the bony fishes, are excellent models to study endocrine control of reproduction.5 Several studies in teleost suggest higher levels of 11-KT as compared to T during all the stages of spermatogenesis, showing the importance of 11-KT for the normal progression of spermatogenesis.6,7 For instance, in the pejerrey, Odontesthes bonariensis, 11-KT is produced in undifferentiated gonads when exposed to masculinizing temperatures before morphological sex differentiation.8 In the previous studies, h11b expression was found to correlate with the testis development as well as with the seasonal cycle and expression of other testis-specific steroidogenic enzyme genes in Clarias gariepinus and C. batrachus. 9,10 In the rainbow trout, Oncorhynchus mykiss, h11b expression was detected well before and during testicular development.11–13 Additionally, H11b is an important enzyme for glucocorticoid and minerelocoticoid production in vertebrates. The biosynthesis of corticosterone and cortisol in the interrenal cells of the kidney of several teleosts and adrenal cortex in mammals also requires H11b.14 The production of corticosterone, cortisol, and OHT is blocked after the treatment of H295 human adrenocortical carcinoma cell line with CYP11B1 inhibitor.2 Studies have also shown that polymorphism in CYP11B1 (H11b analog) and CYP11B2 (aldosterone synthase) genes is associated with primary hyper-aldosteronism and hypertension in human.15,16 In the rainbow trout, O. mykiss, isoforms were known to be resulted from specific genome duplication.11,17 Such evidence has also been reported in mammals.18 A study in catfish reported the existence of four different forms of H11b10 named as regular type, variant 1 (Var1), variant 2 (Var2), and variant 3 (Var3). This also suggested a credible role of H11b considering the changes in 11-KT levels and h11b expression during different developmental phases, together with H11b localization in Sertoli cells in addition to interstitial/Leydig cells.10 However, only the regular form of H11b comprised of all five regions (steroid binding site, Ozol’s and aromatic region, and oxygen and heme-binding sites), which are characteristics for P450 enzymes. The variant forms possessed complete steroid binding site while lacking other regions (Fig. 1), suggesting that these variants might not have any enzyme activity but could provide a scope for steroid binding.10 In another study on similar lines, two isoforms of H11b from the Nile tilapia, Oreochromis niloticus, were cloned, of which isoform-2 was nonfunctional. It was hypothesized that H11b isoform-2 binds to steroids and is solely present to regulate substrate concentration.19 Thus, examining the molecular-level interaction of the substrates with H11b protein and its variants is crucial to understand fish reproduction, more importantly in males.

Sequence alignment of regular form of cytochrome P450 11β-hydroxylase (H11b) against its three different variants, Var1, Var2, and Var3. The symbols star, dot, and dash represent the identical residues, similar residues, and gaps, respectively. The steroid binding, oxygen binding, Ozol’s, and aromatic and heme-binding regions are marked with rectangular boxes.
The present study aims to analyze the difference in the binding affinities of T to the H11b regular form and its variants of C. batrachus using computational methods such as molecular modeling of the H11b and its variants.20–22 H11b was docked against T and molecular dynamics (MD) simulations were performed23–25 to understand its binding mechanisms.26 To achieve this, the three-dimensional structures of H11b and its three variants were generated using homology modeling. The structures were validated, and the probable binding sites of the substrate were identified in all the H11b proteins using docking. Furthermore, MD simulations were carried out to analyze conformational changes in protein, key interactions, and binding affinities of the ligands toward all the variant forms of H11b.
Methods
Modeling the structure of H11b proteins
For homology-based modeling, the best possible template structure was identified using the National Center for Biotechnology Information-Basic Local Alignment Search Tool27 against the structures in protein data bank (PDB). The structure of H11b and its variants were then modeled by the Modeller 9.12 software28 using the identified template. The selected models were further optimized by loop refinement using Modeller 9.12. The quality of the loop-refined models of H11b proteins was validated using Ramachandran plots29 and Z-score.30
Ligand docking
The structure of T was extracted from PDB (ligand id: TES). Charges on the protein and ligand were assigned by the Gasteiger method using MGL-Autodock Tools (https://ccsb.scripps.edu/mgltools/).31 The grid for searching the binding site was selected based on the template protein used for the modeling of H11b. The docking was performed in Autodock Vina.32 Pymol33 and Ligplot34 were used to analyze the interactions.
Molecular dynamics simulation
MD simulations of H11b proteins and their protein–ligand complexes were performed in GROMACS 4.6.535–38 with Gromos54A7 force field. The system to be studied was solvated in a box with simple point charge-extended water molecules. The structures were initially energy minimized. Then, it was equilibrated under constant volume and temperature (NVT) condition followed by constant pressure and temperature (NPT) condition for 200 ps each. Then, production simulation was performed for 20 ns at 300 K and 1 atm pressure. Temperature and pressure were maintained by modified-Berendsen thermostat and Parrinello-Rahman39 type pressure coupling, respectively, with a time constant of 2.0 ps. A cutoff of 1 nm was used for short-range coulombic and Lennard–Jones interactions. Long-range electrostatic interactions were treated by the particle mesh ewald scheme.40 Parallel linear constraint solver algorithm41 was used for bond constraints. Coordinates, velocities, trajectories, and energies were collected at every 2 ps for further analysis. Trajectory analysis such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), solvent-accessible surface area (SASA), and radius of gyration (Rg) were done using the Gromacs modules. Binding energy was calculated for each system by the molecular mechanics-Poisson–Boltzmann surface area (MM-PBSA) method using the g_mmpbsa tool implemented in GROMACS.42,43
Results
Homology modeling of H11b and its variants
The experimentally determined 3D-structure of H11b protein of C. batrachus is not available. Therefore, the structure was modeled using Modeller 9.12. Modeller uses closely associated homologous protein as a template to construct the unknown protein model. The basic principle behind homology-based modeling is that two proteins with high sequence similarity might share a similar structure. Compared to other methods such as threading and ab initio modeling, homology modeling is more reliable due to the structural proximity of the unknown protein with the template.44
The initial search against the proteins available in PDB showed that human cholesterol side-chain cleavage enzyme, CYP11A1 (PDB ID: 3N9Y),45 has the highest similarity of 45.6% and the query coverage of 85%. The CATH domain analysis shows that the protein structure belongs to cytochrome P450 topology. The structure of CYP11A1 is available for the residues only from 41 to 521. The initial 39-residues in the N-terminal are identified to be a signal peptide,46 and it is unstructured. Therefore, the corresponding sequence in the H11b protein was removed after aligning the full-length CYP11A1 against H11b (Fig. 2), and the rest of the sequence (amino acids 51–546) was considered for homology modeling using CYP11A1 as a template. Since H11b is a heme-protein, the structure was modeled with and without heme to analyze the binding of T with both holo and apo forms of the protein. H11b with heme (reg+heme) was modeled using Modeller’s heteroatom transfer protocol by considering the heme as a rigid ligand47 (Fig. 3A). In the template, four arginine (Arg) and two tryptophan (Trp) residues from the polypeptide chain interact with heme through H-bonding, and a cysteine (Cys) residue interacts with iron atom of the heme. In the H11b, all the interactions are conserved except one. Trp is replaced with tyrosine residue (Fig. 3B). Also, the model for apo form of H11b (reg-apo) was developed. The secondary structural contents of the modeled proteins were analyzed using the DSSP-Web server (http://bioinformatica.isa.cnr.it/SUSAN/NAR2/dsspweb.html). The analysis showed that the holo-protein had 51.3% of α-helix and 8.7% of β-sheet, whereas the apo-form contained 52.3% of α-helix and 9.1% of β-sheet. The rest of the regions were turns and coils. The template structure obtained from the PDB contained 50.3% of α-helix and 9.5% of β-sheet, suggesting that the secondary structural contents of the modeled proteins were comparable with the experimentally determined template structure.
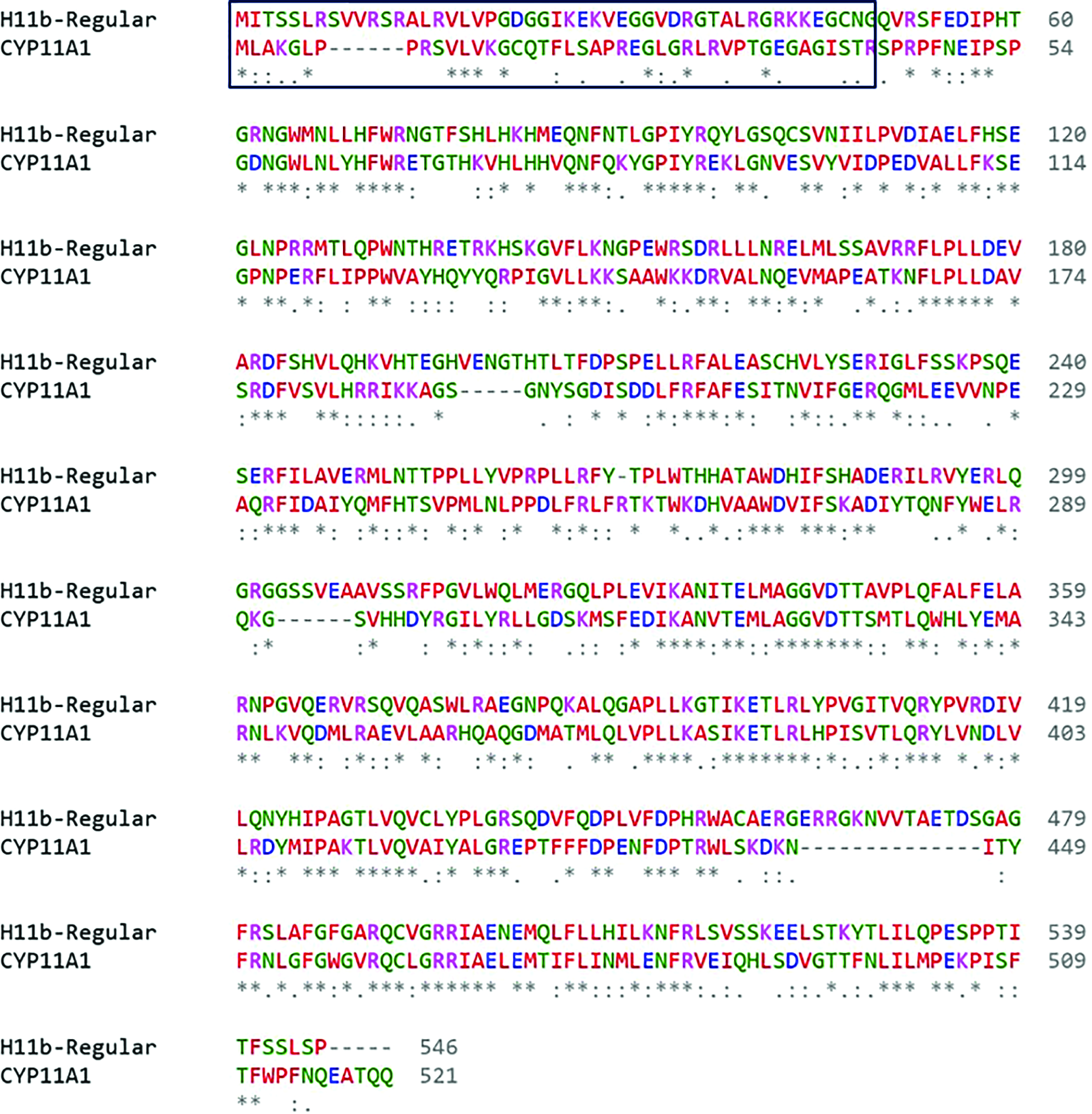
Sequence alignment of regular form of H11b against the template CYP11A1. The symbols star, dot, and dash represent the identical residues, similar residues, and gaps, respectively. The unstructured region, a part of signal peptide, in the N-terminal is marked with rectangular box.

(A) Three-dimensional structure of regular form of H11b with heme modeled using Modeller. The helix, sheet, and loop are represented with cyan, magenta, and brown ribbons, respectively. Heme group is shown in red sticks with the central iron atom as sphere. (B) The residues interacting with heme are highlighted as yellow sticks and labeled.
Furthermore, the variant forms, Var1, Var2, and Var3, were also modeled using the same template (Fig. 4). The similarity and sequence coverage are presented in Table 1. The α-helical contents for the variants Var1, Var2, and Var3 were 57.7%, 60%, and 60%, respectively, and the β-sheet contents were 3.9%, 5%, and 3.9%, respectively. The rest of the regions formed turns and coils in the structure. All five structures were subjected to loop refinement in Modeller. To validate the modeled structures, Ramachandran plot of each model was analyzed (Fig. 5), and Z-score values were also calculated (Table 1). The number of outliers in Ramachandran plots was found to be less than 1% for all the models and mean Z-score was in the range of –0.04 to –0.16. These results suggested that the protein structures are comparable with the experimentally determined structures and could be used for further studies.

Three-dimensional structure of (A) reg-apo, (B) Var1, (C) Var2, and (D) Var3 forms of H11b modeled using the Modeller. The helix, sheet, and loop are represented with cyan, magenta, and brown ribbons, respectively.

Ramachandran plot of the protein models developed for reg+heme, reg-apo, Var1, Var2, and Var3 forms of H11b protein. The darker blue and yellow regions represent the favored regions, and the lighter shades represent the allowed regions, whereas the colorless regions represent the disallowed regions.
Sequence similarity and coverage between template and query*
After removal of the unstructured region/signal peptide in the N-terminal.
Docking of testosterone
T molecule obtained from the PDB database (ligand id TES) was docked against H11b, and its variant forms using Autodock Vina. Autodock Vina is an open-source platform that uses an iterated local search global optimizer algorithm, which is faster and more efficient in identifying the binding orientation.32 The binding site search was constrained to the binding pocket identified from the template. The template, 3N9Y, is a cocrystallized structure with cholesterol that binds to the conserved steroid binding pocket. The best binding poses were chosen based on the highest binding affinity obtained from the docking (Table 2). T shows the binding affinity with the proteins on the order of reg+heme > reg-apo > Var2 > Var3 > Var1. The bound conformations of T against all the variants of H11b are presented in Fig. 6. Analysis of the binding pocket using Ligplot (Fig. 7) indicated that the ligand is mostly surrounded by hydrophobic residues in regular and in all variant forms of H11b. The binding pocket consists of mostly Trp and leucine residues. Also, arginine-77 (Arg77) is also noted in the cases of regular, Var2, and Var3. It may be noted that the cholesterol-binding site in the template is also surrounded by hydrophobic residues such as Trp, phenylalanine, and isoleucine (Ile) along with an Arg residue. This emphasizes the fact that steroid-binding pockets are highly conserved across the bony fishes10 as evident through sequence analysis.

The binding pocket of T with the (A) reg+heme, (B) reg-apo, (C) Var1, (D) Var2, and (E) Var3 forms of H11b obtained from docking. T is represented in ball and stick (gray—carbon, white—hydrogen, and red—oxygen) models. The major interacting residues are presented as green sticks and labeled. All the interacting residues in the binding pocket are shown as 2D-plot in Fig. 7.
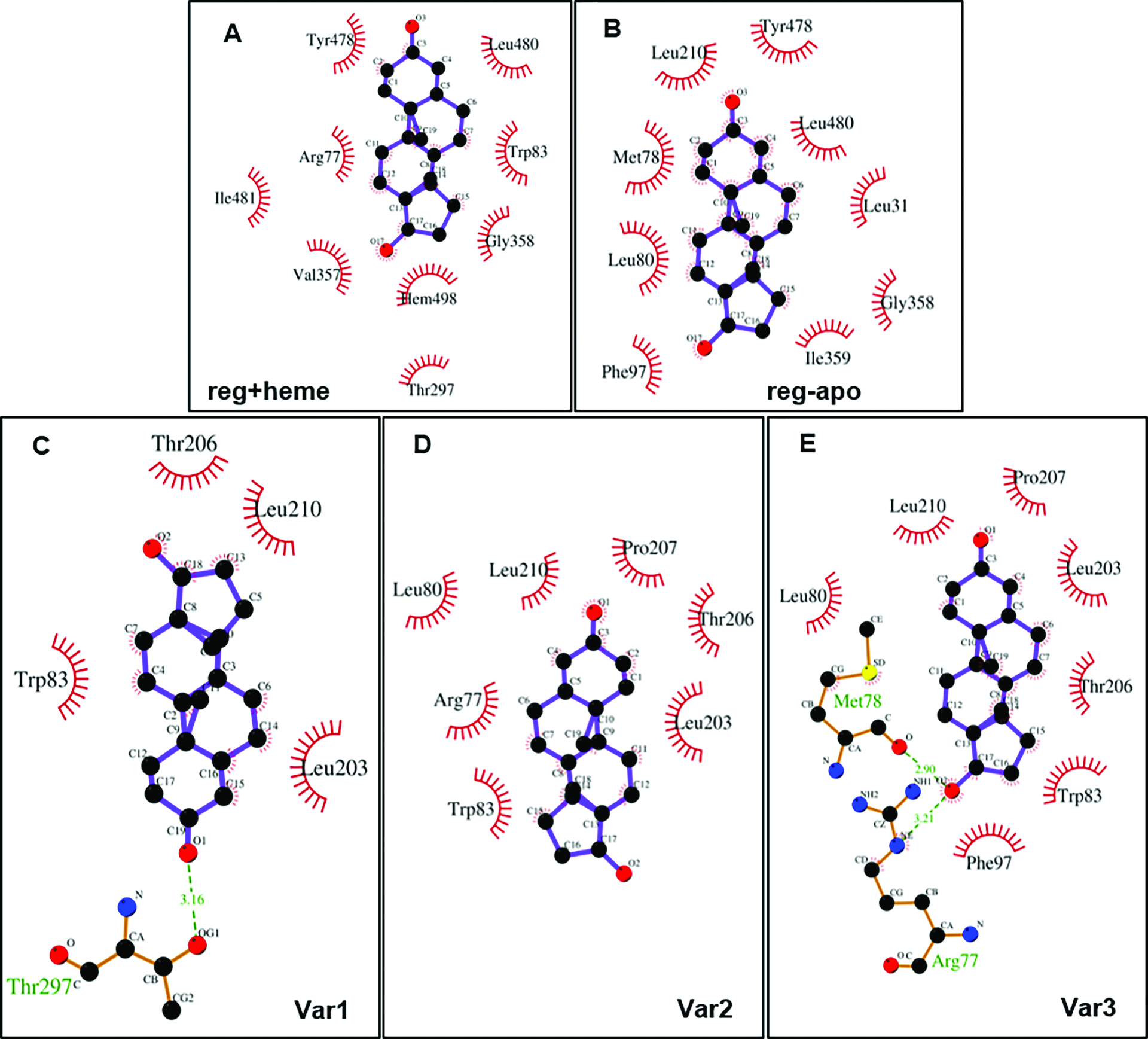
The residues of H11b and its variants interacting with T are shown as a 2D-plot created using Ligplot. The ligand is shown as purple sticks, where black and red balls represent carbon and oxygen atoms, respectively. The red brushes represent the amino acid residues interacted by hydrophobic interactions, whereas green-dotted lines represent H-bonding interactions.
Binding of testosterone with different forms of H11b
Molecular dynamics simulation
To understand the conformational fluctuations and the binding affinity of the ligand, MD simulations of H11b and its variants, and their ligand-bound complexes were performed using the Gromacs software. The Gromacs software is robust for many-atom classical simulations and can perform simulations with macromolecular force fields such as GROMOS with compatible water models. Also, it has tools to analyze the structural changes during the simulation and to calculate interaction energies.48
From the simulations of protein–ligand complexes, RMSD of the whole protein chain and RMSF of each residue from the initial structure were calculated by considering Cα-atoms as reference points (Fig. 8). The regular forms of H11b had the lowest RMSD during the simulation, and among the variants, Var1 and Var2 had larger RMSD values than Var3. Within the regular form, the apo form had slightly larger RMSD, which could be due to the loss of heme moiety. Upon binding of the ligand, T, the RMSD values of reg-apo, Var2, and Var3 were reduced, whereas reg+heme and Var1 showed a marginal increase. From the RMSF analysis (inset in Fig. 8D), it was observed that the increased fluctuations were from the residues, which are distant away from the binding pocket, and, thus, it might not alter the binding sites directly. The secondary structural analysis using DSSP-web tool suggested that α-helix and β-sheet contents of the proteins remained almost the same upon binding of the ligand, whereas fluctuations were observed in the loop regions.
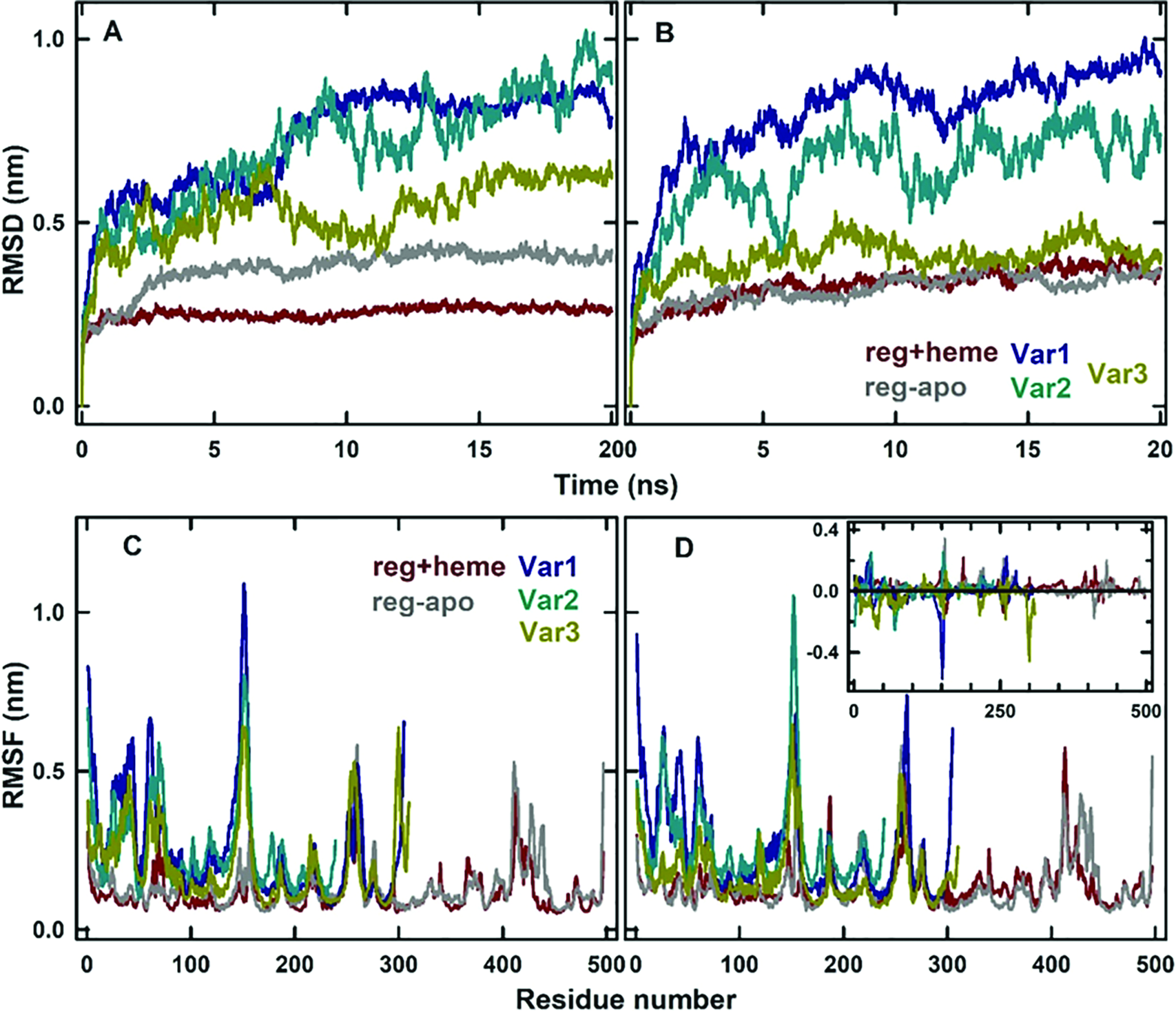
RMSD changes of H11b in reg+heme (red), reg-apo form (gray), Var1 (blue), Var2 (cyan), and Var3 (green) forms in the absence (A) and the presence (B) of ligand T. Similarly, RMSF changes of each residue in different protein forms in the absence (C) and the presence (D) of ligand T. The inset in (D) shows the difference in the RMSF between the ligand-bound form and free form of the proteins.
The changes in Rg during the simulations were calculated (Fig. 9A). The Rg values were more for the regular forms of H11b, since they have more number of residues and are expected to have slightly larger hydrodynamic radii than the variants. The Rg values of the variants slightly decreased at the initial period of MD simulation and attained stable values. The binding of T did not alter the Rg values of regular forms of the protein, and Var2 and Var3; however, the Rg value of Var1 was slightly reduced after 15 ns of the simulation time (Fig. 9B). The SASA values of the regular forms were slightly less than that of the variant forms, and Var2 had the highest SASA. The binding of T did not alter the SASA values of any of the proteins, suggesting that the global conformation of the proteins and their corresponding protein-T complexes are similar.

Rg values of H11b in reg+heme (red), reg-apo form (gray), Var1 (blue), Var2 (cyan), and Var3 (green) forms in the absence (A) and the presence (B) of ligand T. SASA of the different protein forms in the absence (C) and the presence (D) of ligand T.
Protein–testosterone binding energy
The binding interaction between the proteins and T was analyzed using the MM-PBSA method. The method calculates the binding energy based on the electrostatic interactions, van der Waals interactions, polar solvation, and nonpolar solvation energy via SASA energy. The contribution from each of these interactions is reported in Table 2 and shown in Fig. 10. The calculations show that T binds most favorably with the reg-apo form of the protein, which is mostly driven by the excess electrostatic interaction energy. The Var1 and reg+heme forms showed similar binding affinity. Var2 had less electrostatic interaction with the ligand; however, the van der Waals energy along with the lowest polar solvation penalty facilitated the binding of the ligand. Though the electrostatic interaction is higher in Var3, the high penalty of polar solvation energy weakens the binding. The overall results suggest the hydrophobic interaction favors the binding of T with the proteins along with the low penalty of polar solvation.

(A) Binding energy of T with different variants of H11b calculated using the MM-PBSA method. (B) The contribution of individual components, electrostatic (red), van der Walls (gray), polar solvation (pink), and SASA energy (cyan) for the total binding energy.
Discussion
Examining the binding of T with different variants of H11b is essential to understand the regulation of the concentration of T. The protein models of H11b and its variants were developed from a homologous protein and were validated. The binding of T with the regular form H11b with and without heme cofactor was studied. Also, the binding with variant forms was analyzed. The results indicate that T favorably binds to all the forms of the proteins with negative binding free energy. The highest binding could be seen with the reg-apo form of the protein, whereas lowest binding is shown with Var3. The results, thus, suggest that the efficient binding of T with varying binding free energies could play important role in the storage and regulation of T in the cellular system. Based on the homology study, it is predicted that the presence of H11b variants in the species with high similarity yet the variations in the C-terminal region might be due to recent mutations.
Previous studies in catfish indicated that h11b (regular type) has a high identity with Danio rerio and O. niloticus, suggesting common functions of these enzymes in teleosts, wherein indirect role of h11b was depicted in early spermatogenesis in teleosts following a seasonal pattern as evidenced through expression and localization studies.10,11,49,50 However, the detailed role of H11b in spermatogenesis needs further investigation. Additionally, the loss of Ozol’s, aromatic region, and oxygen and hemebinding sites in the variants indicated that the enzymes lack the hydroxylase activity due to the loss of the redox centers, which are present in the heme-binding region corroborating the findings of the current study. Interestingly, the presence of all the domains solely in h11b (regular type) indicates it as an essential enzyme for the production of 11-KT. Another study demonstrated that h11b isoform 2 of O. niloticus lacked important regions. It was hypothesized that the function of h11b isoform 2 (nonfunctional) is to act as a steroid-binding protein to regulate the substrate concentration in the steroid-producing cells.19 Furthermore, reports, in this line, provided in vivo evidence to 11β-hydroxylation of T to produce the 11-KT precursor OHT, which was supported by unchanged concentrations of T in h11b-deficient zebrafish, also indicating that excess androstenedione was not converted to T,51 and this was shown earlier in vitro. 52 However, this has not been analyzed in any other fish model till date.
Conclusion
Catfish, C. batrachus, contains four different forms of H11b, which are known as regular, Var1, Var2, and Var3 forms. The variant forms contain steroid binding site but lack other regions. Thus, they might be used to regulate steroid concentration. The binding affinity of steroid, T with all four forms of H11b, and nonheme form of H11b (apo-form) is studied using computational methods. The variant forms were modeled using homology modeling, and the T binding site was identified by molecular docking. The binding affinities were calculated by the MM-PBSA method using MD simulation. The regular form has a higher binding affinity than variants, and among the variant forms, Var1 has the highest binding affinity. This could be due to favorable van der Waals interactions between the protein and steroid. Furthermore, in vivo and/or in vitro enzyme activity analysis, in this line, is necessary to confirm that the single functional form (regular type) might be involved in the competitive binding of T to produce 11-OHT and, in turn, 11-KT in catfish. In this context, specific cell organelles preparation coupled with enzymatic assay may provide insights on H11b functionality in vitro and in vivo. Furthermore, in vivo localization analysis of H11b in testicular cells will be advantageous.
Footnotes
Acknowledgments
We acknowledge the Bioinformatics Facility and the Centre for Modelling, Simulation, and Design at the University of Hyderabad for computational facility. We thank Dr. Anbazhagan Rajakumar, University of California, San Diego, USA, for providing the sequences used in this study.
Abbreviations
11-hydroxytestosterone 11-ketotestosterone 11β-hydroxylase arginine basic local alignment search tool constant pressure and temperature constant volume and temperature cysteine isoleucine leucine molecular dynamics molecular mechanics Poisson–Boltzmann surface area National Center for Biotechnology Information parallel linear constraint solver particle mesh ewald protein data bank root mean square deviation root mean square fluctuation simple point charge—extended solvent-accessible surface area testosterone tryptophan tyrosine variant 1 variant 2 variant 3