
Abstract
Since the first news about the detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) appeared, a large amount of data on the variability of the virus genome have accumulated. Most of the mutations entrenched in the viral genome are aimed at improving the mechanisms of host cell penetration, altering the degree of binding to protein receptors, evading the immune system, and suppressing the antiviral immune response. Knowledge of the functional role of mutations will allow improving diagnostic methods, treatment, and vaccine prophylaxis regimens, as well as predicting the further spread and evolution of the virus. In this study, we analyzed a number of SARS-CoV-2 virus mutations in the context of viral epitope affinity for most common in Russia HLA class I and class II alleles (according to Allele Frequency Net Database). This study examined clade-forming mutations of viral clades that are classified as variants of concern according to the WHO classification.
We found that some mutations reduce the number of predicted epitopes, the number of HLA alleles that bind them, or the number of both epitopes and HLA alleles simultaneously. Mutations of the viral clade B.1.1.7 (S:Y144del, S:H69-V70del, and S:A570D), mutations of the viral clade B.1.617.2 (S:T19R, S:G12D, S:F157del, and S:R158del), and mutation N:R203K related to all clades except B.1.617.2 have pronounced effects on reducing epitope affinity for HLA alleles, with some of them affecting on epitopes of all the studied strong and weak binders lengths in both HLA class I and class II.
Introduction
The rapid spread of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus is accompanied by a sharp aggravation in the global epidemiological situation. The situation is complicated by changes in the virus genome that increase its pathogenicity and contagiousness. SARS-CoV-2 is a single-stranded RNA-containing virus of the genus Betacoronavirus with a genome size of approximately 30 kb.1 The SARS-CoV-2 genome encodes four structural (nucleocapsid (N), spike (S), membrane (M), and envelope (E)) and 16 nonstructural proteins.2–4 Currently, the rate of evolutionary change in the virus is estimated to be 1 × 10–3 substitutions per site per year, which is equivalent to one change in the genome every 2–2.5 week.5,6 Identification and study of substitutions, and deletions and insertions that affect the properties of the virus are essential for effective immunological monitoring.
The main cells of acquired antiviral immunity are T-lymphocytes. CD8+ T-lymphocytes, in association with HLA class I alleles, recognize foreign antigens and destroy infected cells. CD4+ T-lymphocytes, in complex with HLA class II alleles, recognize viral antigenic determinants located on antigen-presenting cells, participate in the activation of CD8+ T-lymphocytes, and also promote synthesis of specific antibodies by B-lymphocytes by activating them after receiving the corresponding signal.
In this study, we performed bioinformatic analysis of viral epitope and HLA most frequent in Russia alleles affinity. A number of bioinformatics tools currently existto predict epitopes, HLA alleles, and T-cell receptor alleles by amino acid sequence. These algorithms exploit tailored machine learning strategies to integrate different training data types, resulting in state-of-the-art performance and high reliability.7 The algorithms used for predicting the affinity of epitopes and HLA alleles and results of their work alone do not qualify for high research significance, but describe an effective method for calculating the affinity of linear amino acid sequences for HLA classes I and II alleles and studying the immunogenicity of the epitope-MHC complex.
Materials and Methods
SARS-CoV-2 Virus Genome Sequences
The sequence NC_045512.2 downloaded from NCBI8 was used as the reference nucleotide sequence of the SARS-CoV-2 virus genome. The VIGOR4 tool9 was used to translate nucleotide sequences into amino acids and annotate them. The amino acid sequences of structural and nonstructural proteins of the virus with their genomic coordinates, the name of the gene, and its expression product were obtained at the end of VIGOR4 processing.
Analysis of the Prevalence of Viral Clades in Russia
To assess the prevalence of viral clades in Russia, 5,652whole genome sequences of virus isolates were downloaded from the GISAID10 resource for the period from January 1, 2020 to December 10, 2021. Sequences with high coverage (the GISAID filter, whose definition is “only entries with <1% Hs and <0.05% unique amino acid mutations (not seen in other sequences in database) and no insertion/deletion unless verified by submitter”) were selected for analysis to exclude the possibility of sequencing errors.
The bioinformatic tool of the same name (pangolin v3.1.11) was used to determine whether a viral genome sequence belongs to a particular clade according to PANGOLIN nomenclature.11 The whole genome sequences of the virus clades isolated on the territory ofRussia between January 1, 2020 and December 10, 2021 referred to 111 different clades, five of which werethe most common, the number of sequences related to them exceeded 200. The results of the PANGOLIN tool are shown in Figure S1 of the supplementary material.
HLA Alleles
To predict viral epitopes affinity, the most frequent HLA alleles found in Russia and found in the AlleleFrequencies database (WEB database, 2003–2020) were selected.12 The alleles were selected from Russian data corresponding only to the gold population standard, with a resolution of two fields. Alleles were considered frequent if their frequency of occurrence (the average value for all allelic frequencies for a given allele) was greater than or equal to 5%. The selected alleles included five HLA-A alleles, four HLA-B alleles, seven HLA-C alleles, four HLA-DPB1 alleles, seven HLA-DQB1 alleles, and eight HLA-DRB1 alleles. Alleles with a frequency of more than 10% were considered the most common: HLA-A*02:01, HLA-A*03:01, HLA-A*24:02, HLA-B*07:02, HLA-C*03:04, HLA-C*04:01, HLA-C*06:02, and HLA-C*07:02 for class I, and DPB1*02:01, DPB1*04:01, DPB1*04:02, DQB1*02:01, DQB1*03:01, DQB1*03:02, and DQB1*03:03 for classII.
Predicting Viral Epitope Affinity
For each clade, only clade-forming mutations were selected and inserted into the reference amino acid sequence. A pipeline based on pVACtools pipeline was used to predict the affinity of viral epitopes and their binding alleles.13 Two types of protein sequences (wild-type and mutant) flanked by 7, 8, 9, 10, 11, 12, 13, 14, and 15 amino acids for each length from 8 to 16, respectively, on each side relative to the nonsynonymous substitution (except for S:D3L for N protein—flanked by two amino acids on the left) served as the input file for the pipeline.
Epitope size for HLA class I was set in the range of 8–11 amino acids, and 12–16 amino acids for HLA class II. The IC50 threshold value was limited to 50 for strong binders and 500 for weak binders.14 Based on various comparative articles,14–26 the affinity of wild-type and mutant epitopes was predicted using different algorithms: MHCflurry,21 MHCnuggetsI,23 and NetMHCpan15,19 for class I HLA and MHCnuggetsII,23 NNalign,18 and NetMHIICpan for class II HLA. For further filtering, the IC50 BestScore value was used, after which the epitopes found for the HLA class I alleles were further selected by the netCTLpan tool for TAP transport efficiency—those with less than 0, and for proteosome degradation—those with less than 0.5 were selected to ensure that only peptides with a high probability of being produced after proteosome degradation and transport by TAP proteins remained in the analysis. Only those epitopes that contained a position of interest were selected for the final analysis.
Results
Affinity of Epitopes for HLA Alleles Most Common in Russia
The study design is shown in Figure1.
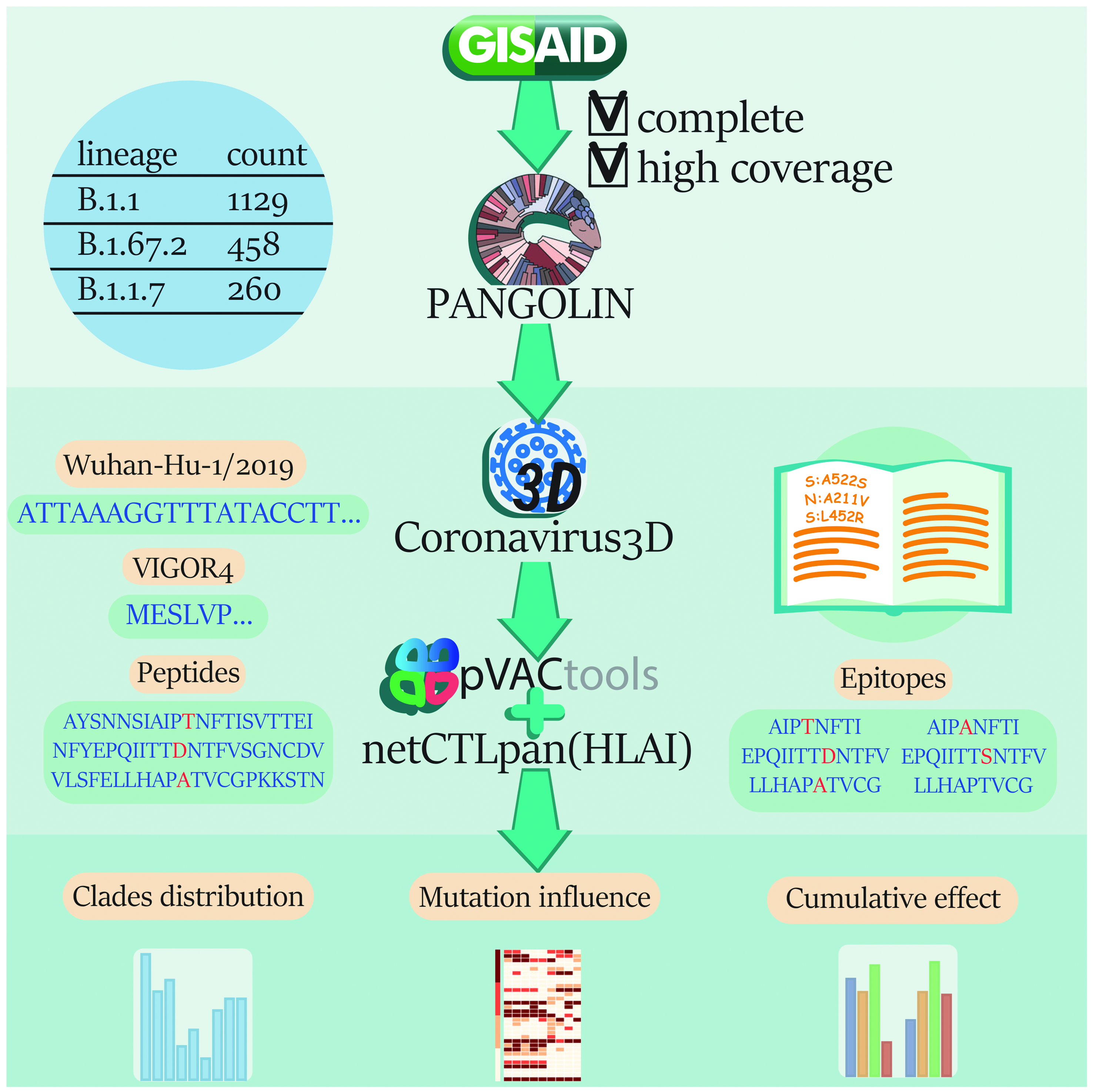
Study design. The pipeline contained the basic steps: downloading of SARS-CoV-2 sequences from GISAID for Russia, annotation of sequences by clades using PANGOLIN, annotation by reference nucleotide sequence using VIGOR4, search for clade-forming mutations on the coronavirus3D resource for the most common clades, generation of wild-type and mutant peptides, prediction of epitopes and HLA allele affinity using algorithms based on pVACtools, and analysis of the obtained results: clades distribution, mutation influence, and cumulative effect of all mutations in a clade.
Having studied the distribution pattern of the SARS-CoV-2 virus clades in Russia, we found that the largest part of the viral sequences isolated and sequenced in Russia between January 1, 2020 and December 10, 2021 belong to the clades B.1.1, B1.1.7, B.1.1.317, B.1.1.523, and B.1.617.2: Figure S1 of the supplementary material. It is important to note that clades B.1.617.2 and B.1.1.7 are classified as variants of concern by the WHO. Mutations of the spike (S) and nucleocapsid (N) proteins of the virus that are entrenched in the viral genome were selected for further analysis. In this study, 31 S protein mutations and 10 N protein mutations were analyzed. The full list of the studied mutations is presented in Table1.
List of mutations analyzed in this study. The position of interest, which is flanked by 10 amino acids on both sides as example, is highlighted in red in peptide column. Mutations from Ref. 27
After the degree of binding of viral epitopes of lengths 8, 9, 10, and 11 amino acids for HLA class I alleles and 12, 13, 14, 15, and 16 amino acids for HLA class II alleles was predicted for selected wild-type and mutant peptides, we calculated the number of predicted viral epitopes for mutant and wild-type sequences and the number of HLA alleles binding them, to assess how certain mutations affect the degree of affinity of viral epitopes for MHC. Comparison of these rates among themselves for each individual length and for all lengths cumulatively showed that some of the considered mutations significantly reduce the affinity of possible for presentation epitopes, i.e., epitopes which could be presented on cell membrane.
SARS-CoV-2 N Protein Mutations Have Less Impact on Epitope Affinity for HLA Alleles than S Protein Mutations
The results of comparing the predicted affinity level of viral epitopes (binders) and HLA classes I and II alleles are shown in Figure2. For HLA class I, the S:Y144del mutation (B.1.1.7) reduced affinity of both strong (IC50≤ 50 nM) and weak binders (50 < IC50 ≤ 500nM) of all investigated lengths. The S:A570D mutation (B.1.1.7) reduced affinity of strong binders of all studied lengths. S:E156del (B.1.1.523) and S:R158del (B.1.617.2) mutations affected all-length HLA class I weak binders affinity. In addition, the studied mutations had an effect on the level of epitopes affinity for HLA class II alleles. The S:Y144del, S:A570D (B.1.1.7), and S:R158del (B.1.617.2) mutations retained influence only on the affinity of weak binders of all lengths studied. S:A845S (B.1.1.317), S:F157del (B.1.617.2), S:G412D (B.1.617.2), S:S477N (B.1.1.317), S:S494P (B.1.1.523), S:S982A (B.1.1.7), S:T19R (B.1.617.2), and S:T478K (B.1.617.2) mutations influenced the affinity degree of weak binders of all considered lengths. Finally, the S:H69-V70del (B.1.1.7) mutation affected the affinity of both strong and weak binders of all lengths.

Effect of SARS-CoV-2 spike protein mutations on epitopes and alleles affinity. (A) Strong binders and (B) weak binders. Lengths of 8–11 amino acids illustrate the effect of mutations on the affinity of epitopes and HLA class I alleles; lengths of 12–16 amino acids illustrate the effect of mutations on the affinity of epitopes and HLA class II alleles.
Only one N:R203K mutation (B.1.1.7) affected strong binders affinity levels at all lengths for HLA class I alleles, and results are shown in Figure3 for both HLA classes I and II. The analysis clearly demonstrates the fact that SARS-CoV-2 S protein mutations have a greater effect on the affinity of epitopes for HLA classes I and II alleles than N protein mutations of the virus.

Effect of SARS-CoV-2 nucleocapsid protein mutations on epitopes and alleles affinity. (A) Strong binders and (B) weak binders. Lengths of 8–11 amino acids illustrate the effect of mutations on the affinity of epitopes for HLA class I alleles; lengths of 12–16 amino acids illustrate the effect of mutations on the epitopes affinity for HLA class II alleles.
Cumulative Effect of S and N Protein SARS-CoV-2 Mutations
To assess the cumulative effect of mutations, we identified five possible mutation effects for each viral epitope length: (1) mutation reduces the number of binding alleles, (2) mutation reduces the number of possible epitopes, (3) mutation reduces the number of binding alleles and possible epitopes, (4) mutation completely limits the affinity of epitopes and alleles (no prediction of binding for mutant epitope compared to wild-type), (5) mutation increases the BestScore value showing the affinity of epitope, i.e., attenuates the affinity of epitopes for the alleles under consideration. If the mutation had influence 5, it was assigned a value of “0.5” in the cell corresponding to this option of influence. Variants 1 and 2 were assigned a value of “1”, variant 3 was assigned a value of “2” as it is a combination of variants 1 and 2, and variant 4 (mutation completely limits the affinity of epitopes and alleles) was assigned a value of “3”, since this type of influence was considered to be the most significant. For each mutation on the same length of the viral epitope, values “1” and “0.5” are possible (e.g., a decrease in the number of epitopes and an increase in the BestScore value), or values “2” and “0.5” (decrease in the number of epitopes and alleles together and an increase in the BestScore value), or one “3” value. Subsequently, the obtained values were summed up for each mutation across all viral epitope lengths, and then the values of all mutations belonging to the same clade were summed up, which was an indicator of the cumulative effect. Since a total of nine epitope lengths were involved in the analysis, the maximum cumulative effect after summing over the lengths could be 18. Furthermore, depending on the number of mutations in each clade, the maximum value could be 54 (B.1.1), 162 (B.1.1.317), 234 (B.1.1.523, B.1.1.7), and 252 (B.1.617.2). In Figure4, the results of the assessment of the mutations cumulative effect are presented, which demonstrate that the effect increases with an increase in the number of clade-forming mutations. This inference is correct for the strong class I binders (blue bar), the weak class I (orange bar), and II (green bar) binders in all five clades. In addition, the study demonstrates a lower cumulative influence score for the strong class II binders group (red bar) in all clades.
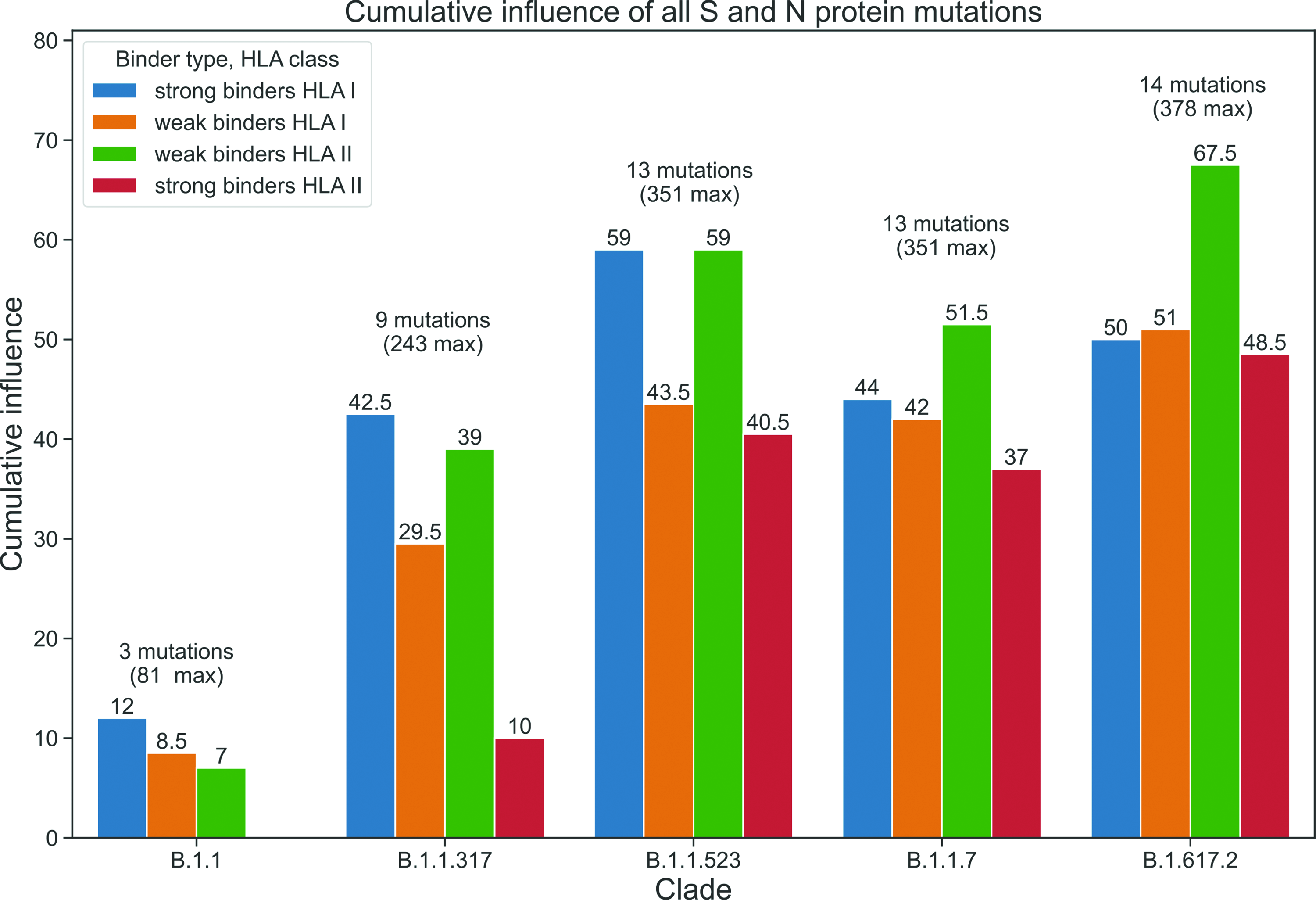
Cumulative effect of S and N protein mutations of each common in Russia clade.
Mutations of the N and S Proteins of SARS-CoV-2 Limit Epitope Binding to Some Common HLA Classes I and II Alleles in Russia
Some of the S-protein mutations considered in this study limited epitope binding (mutant epitope does not bind this allele within the studied IC50) to the most common (AF ≥ 10%) HLA class I alleles in Russia (Figure5), Table S1 for strong binders and Figure S2 and Table S2 for weak binders, as well as HLA class II alleles (Figure5), Table S3 for strong binders and Figure S2 and Table S4 for weak binders.

Clade-forming mutations of the SARS-CoV-2 spike protein that limit epitope binding (mutant epitope does not bind this allele within the studied IC50) to common HLA alleles (AF ≥ 10%), only strong binders. The dotted line marks the boundary of the HLA classes: epitope lengths 8–11—HLA class I alleles and 12–16—HLA class II alleles.
The results of the analysis of the N protein mutations effect on the degree of epitope and HLA allele binding are shown in Tables S5 and S6 of the supplementary material for HLA class I, and Table S7 for HLA class II, and also in Figure6 for strong binders and in Figure S3 for weak binders HLA classes I and II. The mutations under consideration reduced the degree of binding of strong and weak binders to a number of the most common HLA classes I and II alleles in Russia.
Clade-forming mutations of the SARS-CoV-2 nucleocapsid protein that limit epitope binding (mutant epitope does not bind this allele within the studied IC50) to common HLA alleles (AF ≥ 10%), only strong binders. The dotted line marks the boundary of the HLA classes: epitope lengths 8–11—HLA class I alleles and 12–16—HLA class II alleles.
Mutations of Clades that Are not Widespread in Russia Have Less Influence on the Level of Affinity of Epitopes for HLA Alleles Compared to Mutations of Common Clades
Five most uncommon clades in Russia were selected for analysis (TableS8): B.1.1.533, B.1.513, B.1.1.243, B.1.1.251, B.1.469, and 10 clades not included in the five most common but immediately following them in terms of prevalence (FigureS1): B.1.1.397, B.1.1.141, B.1.1.525, B.1.1.294, B.1.1.163, B.1.1.129, B.1.1.336, B.1.1.372, B.1.1.435, and B.1.1.349 (clades B.1 and AY.12 were not included in the analysis because B.1 corresponds to B.1.1 and AY.12 to B.1.617.2 and have same mutations). For the clade-forming mutations of the aforementioned clades, the same analysis was performed as for the mutations in the most common clades, and Table S8 contains a list of mutations for the considered clades.
Cumulative effect for all noncommon in Russia clades is less than this effect for all clades (except original B.1.1) common in Russia (Figure7).

Cumulative effect of S and N protein mutations of each noncommon in Russia clade. Clade B.1.513 has 1 mutation (27 max cumulative effect), clades B.1.1.294, B.1.1.372, B.1.469, and B.1.533 have three mutations (81 max cumulative effect), clades B.1.1.129, B.1.1.163, B.1.1.243, B.1.1.251, and B.1.1.336 have four mutations (108 max cumulative effect), clades B.1.1.141, B.1.1.349, and B.1.1.435 have five mutations (135 max cumulative effect), clade B.1.1.397 has six mutations (162 max cumulative effect), and clade B.1.1.525 has seven mutations (189 max cumulative effect).
Discussion
The pandemic of the new coronavirus infection has challenged the global and domestic healthcare system. The urgent tasks of introducing high-quality methods of diagnosis, treatment, vaccine prophylaxis, and assessment of postvaccine immune response required the healthcare system to use proven tools and develop new ones. The described method for predicting epitopes and assessing the immunogenicity of the epitope-MHC complex for mutant and wild-type peptides allows us to quickly assess the level of changes in the affinity of epitopes and HLA alleles and can be used to predict the rate of spread viral clades and search for candidate epitopes for synthesis of peptide vaccines.
With the emergence of evidence of genome high-rate variation in the SARS-CoV-2 virus, researchers around the world began to investigate the functional role of these mutations. Much of the research has focused on studying changes in the structure of receptor-binding domains and domains that bind to specific neutralizing antibodies.28,29
Our study demonstrated complex use of open-sourced tools for analyzing SARS-CoV-2 mutations influence in the context of epitope affinity with HLA alleles and showed that widespread SARS-CoV-2 clades have higher pathogenicity and contagiousness, which may be related to mutations that affect the degree of viral epitopes and HLA allele affinity. Among the mutations analyzed in this study, viral clade B.1.1.7 mutations (S:Y144del, S:H69-V70del, and S:A570D), viral clade B.1.617.2 mutations (S:T19R, S:G142D, S:F157del, and S:R158del), and the N:R203K mutation related to all but B.1.617.2 clades had the greatest effect on changes in viral epitopes affinity for HLA alleles. In addition, the cumulative effect of S and N protein mutations turned out to be maximal for clade B.1.617.2, which has the greatest number of clade-forming mutations. In studies related to the genetic variability of SARS-CoV-2, most mutations are described as the most beneficial for the virus because they increase host susceptibility, allow to evade the immune response, and contribute to a reduction in the effectiveness of the antiviral immune response.30,31
In addition, there are studies that have shown that SARS-CoV-2 mutations, particularly S protein mutations, minimize the protective effect of existing vaccine,32 which, among other things, are aimed at activating T-cell immune response, although the WHO is quick to assure that at least some protection against new viral clades is available.33
In this study, we have shown that S protein mutations have a greater effect on epitopes affinity for HLA alleles than N protein mutations. Viruses belonging to B.1.1, B.1.1.7, B.1.1.317, B.1.1.523, and B.1.617.2 clades contain S protein mutations in their genome, which significantly affect the affinity degree of epitopes and the most common in Russia HLA alleles, which allowed these clades to spread widely in Russia.
Currently, studies on the functional role of SARS-CoV-2 genome mutations are ongoing, of which special attention is paid to those that have undergone evolutionary selection and are entrenched in the viral genome. The described method for studying mutations in the context of epitope affinity for the HLA alleles makes it possible to study immunogenicity of the epitope-MHC complex and the effect of structural and nonstructural protein mutations on it.
Conclusion
Prediction showed for SARS-CoV-2 most common in Russia clades that the more clade has mutations, the more influence on presumed mutant epitope binding with HLA alleles.