
Abstract
Background:
Breast cancer has been the most recurrent frequent cause of cancer-related deaths in women in India and all around the world. The treatment options have been reportedly more effective in terms of cure at the initial stages of diagnosis, while at the advanced stages, the treatment still remains a great clinical challenge. Many targeted therapies have been reported earlier, which have given impetus to find novel therapies that may lead to increased survival rates in breast cancer patients.
Methodology:
The available literature using PubMed, Scopus and Google Scholar database was thoroughly reviewed using the keywords: breast cancer; P13K/AKT/mTOR; inhibitors; therapeutics; biomarkers and diagnosis. This narrative review of all the relevant papers with significant citations leads the authors to greater insight into the potential therapeutics against breast cancer.
Results:
In most breast cancers, the essential phosphoinositide-3-kinase/AKT/ mammalian rapamycin target (PI3K/AKT/mTOR) signalling pathway has reportedly been triggered, leading to excessive cell growth and tumor formation. The targeting of this important pathway is, therefore, an area of wide-clinical interest, particularly in the breast cancer treatment. Taking these details into account, the current review has reviewed preclinical and clinical studies where a variety of PI3K/Akt/mTOR pathway inhibitors (acting at different levels) have been used either alone or in conjunction with other cancer treatment agents.
Conclusion:
This review includes a detailed understanding of the structure and regulation/deregulation of mTOR, in addition to targeted therapies and the role of mTOR inhibitory drugs in the treatment of breast cancer, along with first and second generations PI3K/AKT/mTOR pathway inhibitors
Introduction
The mammalian target of rapamycin (mTOR) is a serine/threonine (S/T) protein kinase, which plays a key role in various components in the cell, such as cell growth, proliferation, and metabolism by signaling through protein complexes, mTORC1, and mTORC2. mTOR complex 1/2 (mTORC1/2) is evolutionarily conserved from yeast to mammals. The advent of (mTOR inhibitors represented a crucial step in the treatment of different forms of cancer, including breast cancer.1,2 These inhibitors are known to inhibit serine/threonine-specific protein kinase that belongs to the phosphatidylinositol-3 kinase (PI3K) and related kinases (PIKKs) family.3
Oncogenic activation of the PI3K/AKT/mTOR pathway has been known to occur through a variety of mechanisms often including mutation and/or amplification of genes encoding receptor tyrosine kinases (RTKs), e.g., HER2 (ERBB2) and epidermal growth factor receptor (EGFR) (ERBB1), subunits of PI3K (e.g., p110β, p110α, p85β, and p85α encoded by PIK3CB, PIK3CA, PIK3R2, and PIK3R1, respectively), AKT (AKT1), or activating isoforms of rat sarcoma (RAS). Loss of expression or function of phosphatase and tensin homologue (PTEN) through deletions, mutations, or epigenetic silencing is also quite common.4 Rapamycin, a macrolide, is reported to be produced by the microorganism, Streptomyces hygroscopius, which is known to have anticancer, antifungal, and immunosuppressant properties.5,6 Rapamycin derivatives such as rapalogs were further developed as first generation mTOR inhibitors, having similar therapeutic effects in a range of preclinical models as rapamycin, but with improved hydrophilicity and additionally, they could be used for oral and intravenous administrations.7,8 However, rapalogs were not sufficient for achieving broad and robust anticancer effects due to partial mTOR inhibition.9,10 More so, the inhibition of mTORC1 by rapalogs failed to suppress a negative feedback loop that results in phosphorylation and the activation of AKT.11,12 Due to these limitations, the second generation of adenosine triphosphate (ATP)-competitive mTORC1/mTORC2 dual inhibitors was developed. They inhibit mTORC1 and mTORC2 kinase-dependent functions and, thus, block the feedback activation of PI3K/AKT signaling, unlike rapalogs that target mTORC1 only.13,14
In addition, some naturally occurring compounds have been found to down regulate mTOR signaling.15 Keeping in view of the above facts, the role of PI3K/AKT/mTOR pathway in the pathogenesis of breast cancer and preclinical and in vitro findings with respect to PI3K/AKT/mTOR inhibitors have been discussed so as to develop such inhibitors as potential biomarkers against breast cancer.16,17
Structure Preview into mTOR
mTORC1 and mTORC2 are the two structurally and functionally different complexes of mTOR, which play very important role in the pathway at various levels.
mTORC1
mTORC1 consists mainly of five components: raptor (mTOR regulatory-associated protein), mammalian lethal with Sec13 protein 8 (mLST8), 40 kDa proline-rich AKT substrate (PRAS40), Deptor (DEP-domain-containing mTOR-interacting protein), and mTOR.18
Raptor enrolls substrates for mTOR regulating the complex formation. mTOR is the catalytic subunit of mTORC1.19 mTORC1 is negatively regulated by both PRAS40 and Deptor. Enrollment of both PRAS40 and DEPTOR to mTORC1 upholds the reticence of the complex when there is reduction in mTORC1 activity.20 PRAS40 directly inhibits mTORC1 kinase activity by binding to the substrate.21 Activation leads to phosphorylation of PRAS40 and Deptor by mTORC1, which shows a reduced physical interaction with mTORC1 and further encourages mTORC1 signaling.22,23 mTORC1 acts as an energy and a nutrient sensor and also reported to be involved in the regulation of biogenesis, lipid synthesis, autophagy, mitochondrial metabolism, etc.24 Also various amino acids, serum, insulin, and other growth factors are known to forbid the activity of mTORC1 ( Fig. 1A).25,26

The mTOR pathway complexity along with its domain structure. There are mainly six functional components of mTOR domain: HEAT domain (N terminus consisting of 20 tandem HEAT repeats, along with 1,350 residues of mTOR), FAT domain (650-residue of the protein structure), FRB domain (inhibitory action of rapamycin on raptor-bound mTOR), PIKK domain, repressor domain, and FATC domain. At the C-terminus, one part of mTOR consists of kinase domain, which is also known as catalytic domain.
mTORC2
mTORC2 complex comprises six different proteins: rapamycin-insensitive companion of mTOR (Rictor), Protor-1, mSIN1, mLST8, Deptor, and mTOR. Some of the major components, MLST8, Deptor, and mTOR, are usually shared between both mTORC1/C2 complexes. mSIN1 and Rictor are known to stabilize one other. Rictor interacts with Protor-1 as well. Like in mTORC1, Deptor monitors mTORC2 activity negatively.27 mLST8 is essential for mTORC2 function, as knockout of this protein specifically reduces the stability and the activity of this complex.28,29 mTORC2 is a critical regulator of the cytoskeleton through its stimulation of Rac1, Cdc42, paxillin, F-actin stress fibers, and RhoA.30–32 mTORC2 phosphorylates the serine/threonine protein kinase Akt/protein kinase B (PKB) at the serine residue S473, thereby affecting metabolism and survival1,28,33 (Fig. 1B).
The domain structure of mTOR comprises six functional domains: (1) HEAT domain (Huntington elongation factor 3, which is a subunit of protein phosphatase 2A and TOR1), and it mediates protein–protein interactions; (2) FAT C-terminal focal adhesion targeting domain (FRAP-ATM-TRAPP); (3) FRB (a small domain derived from full length mTOR) (FKBP12-rapamycin binding) domain that inhibits rapamycin action on raptor-bound mTOR; (4) PIKK domain; (5) RD (repressor domain); and (6) the carboxy-terminal FRAP, ATM, TRAPP C-terminal domain (FATC) domain29,34,35 (Fig. 1C).
The N terminus consists of 20 tandem HEAT repeats, and C-terminal half of mTOR contains the kinase domain (catalytic domain). The FRB and FAT domains are present in upstream of the catalytic domain. The first 1,350 residues of mTOR consist exclusively of HEAT repeats followed by the 650-residue FAT domain that comprises numerous tetratricopeptide repeats.36 Both types of repeats form extended superhelical domains essential for protein–protein interactions. The FAT domain consists of 28 alpha helices arranged in repeats, which forms a “C”-shaped solenoid around the kinase domain.21,37 The FRB domain consists of 100 residues present in between FAT and kinase domain, and this domain is required for binding to RAPTOR and RICTOR.38 The kinase domain comprises 550 residues and has two lobes: N-terminal and C-terminal lobes with a cleft in between these lobes bound to ATP. The C-lobe forms the binding site for mLST8. The FATC domain is located at C-terminus and comprises 35 residues.21,39 The repressor domain is present in between the kinase and FATC domain, and the deletion of this domain causes the activation of mTOR.21,38,40
Essence of PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway is a significant regulator of cell metabolism, growth, proliferation, and survival during the cell cycle. The pathway is activated by a variety of cellular processes, including angiogenesis, tumor formation, and so on ( Fig. 2). The PI3K/AKT complex activates mTORC1 and is inhibited by the tuberous sclerosis complexes 1/2 (TSC1/TSC2) complex,41 whereas growth factors activate mTORC2.42 mTORC1 controls ribosomal formation and protein synthesis by phosphorylation and inactivating the mRNA translation repressor 4EBP1, as well as phosphorylation and activating S6K.43,44

PI3K/AKT/mTOR intracellular signaling pathway playing a significant role in regulating the cell cycle. The PI3K/AKT/mTOR pathway is activated by the enzymatic receptors having importance in cellular proliferation, cell differentiation, apoptosis, and angiogenesis as well.
A wide variety of growth factors, hormones, and nutrients along with kinases such as class I P13K and AKT are known to activate the mTORC1-mediated signaling.45 The amino acids in the presence of mTORC1 may induce the phosphorylation of S6K1 and 4EBP1 molecules. In contrast, upon amino acids depletion may result in rapid dephosphorylation of above molecules.45 Moreover, the energy level of cells influences the overall activity of mTOR as well. In the moment, the AMP-activated protein kinase levels become downregulated, and the AMPK-mediated phosphorylation of TSC2 further suppresses the mTOR1 activity. However, TSC1/TSC2 complex is not necessarily required for the regulation of mTORC1; instead, the Rheb-GTP is crucial for such regulation. Amino acids are also known to influence mTORC1 activity via human vacuolar protein sorting-34 (hVps34) and class III PI3K as well.46 Moreover, RAS/MAPK (a mitogen-activated protein kinase) signaling also induces mTORC1 signaling as RAS proteins serve as a guanosine diphosphate/ fuanosine triphosphate (GDP/GTP)-control switch. In a typical inactive cell, RAS is coupled to GDP, while in the presence of growth factors, hormones, or cytokines, the activated form of GTP-bound RAS binds to and activates RAF kinase. RAF phosphorylates and activates mitogen-activated protein kinase (MEK), which promotes the extracellular-signal-regulated kinase/ribosomal S6 kinase (ERK/RSK) pathway and affects gene expression, cytoskeleton, and metabolic remodeling. ERK and RSK result in the blockage of TSC2 phosphorylation, which increases TSC1/TSC2 dissociation and further activates mTORC1.47 Inhibitor of nuclear factor B kinase β (IKKβ) is also reported to phosphorylate TSC1, preventing TSC1/TSC2 complex formation and activating mTORC1. Tumor necrosis factor-alpha is another cytokine resulting in the stimulation of mTORC1 activity by triggering AKT. Similarly, mTORC2 phosphorylates AKT in the presence of growth factors.7 Mutations and/or amplification in PI3K/AKT and increased expression or establishment of growth factor receptors such as insulin-like growth factor receptor (IGFR) and human epidermal growth factor receptor 2 (IGFR) could be attributed for the disruption of the PI3K/AKT/mTOR signaling.7,45 PTEN is another negative regulator of PI3K, the functioning of which can be disrupted by mutations, methylation, protein instability, loss of heterozygosity, and abnormal production of microRNA. There are some effector molecules such as S6K1, 4EBP1, and eukaryotic translation initiation factor 4E (eIF4E), which are involved in cellular transformation, and their elevated expression has been linked to bad prognosis in cancer patients. Amplification/mutation of PI3K and increased expression of S6K, AKT, eIF4E, and 4EBP1 may cause several underlying effects such as loss of PTEN function and dysregulation of the mTOR pathway.48
The binding of an extracellular ligand to RTKs is known to stimulate PI3K at the plasma membrane level (Fig. 2). The activated PI3K further leads to the production of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) known to act as a second messenger, which further binds to the pH domain of AKT, resulting in phosphorylation of T308 and S473 with the help of mTORC2 and PDK1. A tumor suppressor gene known as PTEN is a negative regulator of the pathway, any alterations to PTEN due to regulators misbalance oncogenic pathways including PI3K-AKT pathway. Another major component of the pathway is mTORC2, which has important implications in controlling and maintaining the actin cytoskeleton. This component of the pathway has the capability of cell regulation, proliferation, and survival along with cell migration and cytoskeletal remodeling. Overall, the activation of mTORC1 is initiated by some of the major factors like oxidative stress, amino-acid levels, etc. This complex promotes inflammation by skewing the T-cell development. There are many possible ways through which we can arrest mTORC1. The generation of best possible ligands through de novo generation can target the pathway (FRB Domain) further inhibiting the over activation (Fig. 2). Diverse classes of PI3K/AKT/mTOR inhibitors and their therapeutic potential have been elucidated in Table 1.
Diverse classes of PI3K/AKT/mTOR inhibitors/drugs and their therapeutic potential
Moreover, mTORC1 regulates a number of proteins, including CLIP-170 (cytoplasm linker protein-170), HIF-1α (hypoxia-inducible factor 1α), eukaryotic elongation factor 2 kinase, lipin, ornithine decarboxylase, glycogen synthase, PKCδ and PKCɛ, protein phosphatase 2A, p21Cip1 and p27Kip1 cyclin-dependent kinase inhibitors, retinoblastoma protein, and activator of transcription-3.49 mTORC2 controls the function of the small GTPases Rac and Rho by phosphorylating AKT, PKC-, and paxillin (focal adhesion-associated adaptor protein).50–53
According to studies, mTOR is a significant component of cell growth, metabolism, and proliferation. Various growth factors, ATP, amino acids, and oxygen levels are known to effectively regulate the pathway. When the AKT is phosphorylated, it triggers the mTORC1 signaling. PtdIns (3,4,5) P3 is the second messenger formed by class I PI3K, which also binds to the pleckstrin-homology (PH) domain of AKT and PDK1. The kinase is carried to the cell membrane by PtdIns (3,4,5) P3 binding to the PH domain of AKT, which is formed by phosphorylation of PDK1 at Thr308 and phosphorylation of mTORC2 at Ser473. PTEN inhibits AKT activation by transforming PtdIns (3,4,5) P3 to PtdIns (4,5) P2, resulting in decreased AKT recruitment to the cell membrane. AKT is known to be activated by FOXO (members of the class O of forkhead box transcription factors) transcription factors, GSK3 and TSC2, as well as a variety of downstream substrates. Phosphorylation of TSC2 prevents the formation of TSC1/TSC2 complexes, which induce the small GTPase Rheb into the GTP-bound active state further leading to the activation of mTORC1 at Ser2448. mTORC1 is negatively regulated by turning around its activation by Rheb after AKT phosphorylates and inhibits PRAS40.54,55 Both S6K1 and 4EBP1 get phosphorylated by mTORC1, which is activated by interrelating raptor and a TOR signaling (TOS) motif in S6K and 4EBP. mTOR-insensitive signaling pathways such as PDK1, MAPK, and stress-activated protein kinase (SAPK) also activate S6K1 (stress-activated protein kinase).56–58 Despite this, S6K1 phosphorylation is necessary for its activation via mTORC1, and the phosphorylation sites of S6K1 are known to be hindered by mTOR inhibitors. mTORC1 (activated) phosphorylates S6K1, which then phosphorylates 40S ribosomal protein S6, increasing the translation of mRNAs with a 5′-terminal oligo-poly-pyrimidine.59–61 Ribosomal proteins, elongation factors, and insulin growth factor 2 are all targets of S6K1. 4EBP1 binds to and deactivates eIF4E and deactivates it and, therefore, prevents the initiation of protein translation. mTORC1 phosphorylates 4EBP1 at multiple sites, allowing eIF4E to dissociate from 4EBP1, and thus alleviating 4EBP1 inhibition of 4EBP1 on eIF4E-dependent translation beginning.62–64
In breast cancer, RTKs assimilation establishes sensitivity to PI3Kα-selective inhibitors. The overexpression of RTKs (also along with c-MET (mesenchymal epithelial transition factor (MET)), fibroblast growth factor receptors (FGFR), human epidermal growth factor receptor 3 (HER3), and EGFR) reestablished ERK phosphorylation, and the feasibility of the cell concealed by BYL719. This further suggested the proper and authenticated function of RTKs in cell proliferation and signaling. More concisely, in c-MET and EGFR, the overexpression of both of these molecules showed resistance, but on the contrary to this, the overactivation of IGF-1R and/or HER2 showed some sensitivity to BYL719 in breast cancer cells.65,66 The RTKs with an effective and safety features can be probably considered to be as a predictive biomarker for the effectiveness of PI3Kα inhibitors.67 Another study reported an inhibitor DYRK2, whose uprooted expression encouraged Thr631 phosphorylation for the degradation of mTOR. Tyrosine phosphorylation regulated kinase 2 has also been proposed to be used as a potential prognostic marker.68 The general action mechanism of the PI3K/AKT/mTOR inhibitors has been proposed in Fig. 3.13,69
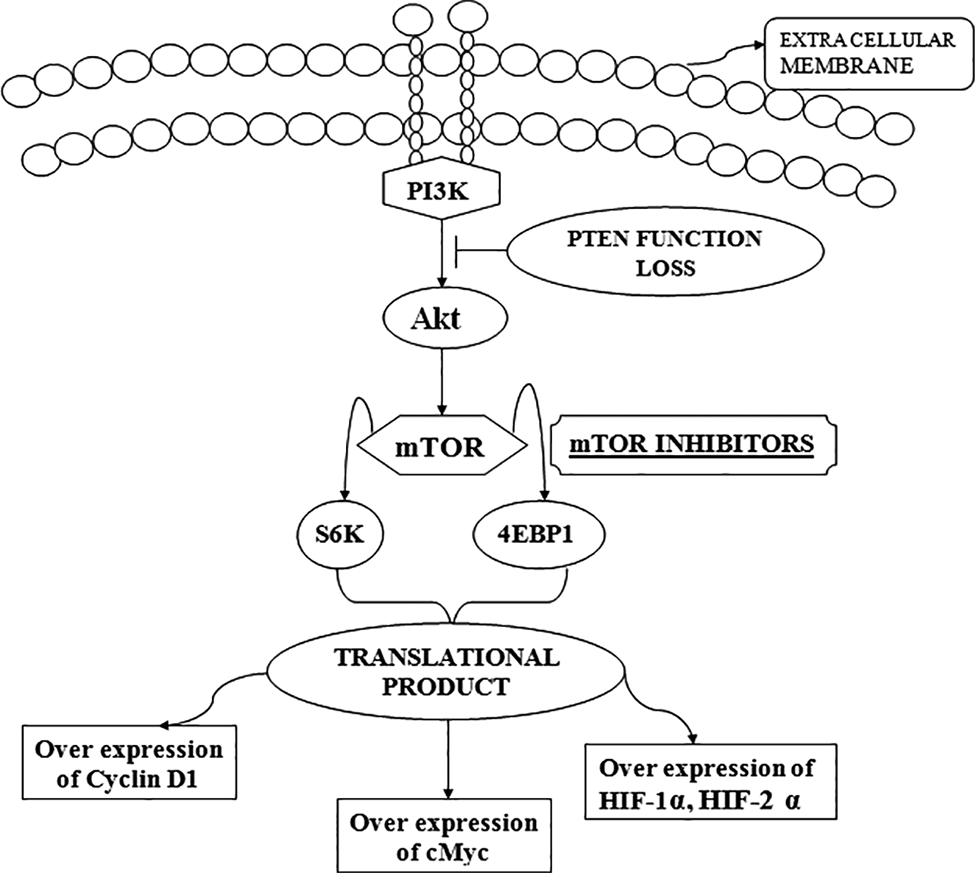
Proposed action mechanism of PI3K/AKT/mTOR inhibitors. Briefly, PI3K inhibitors are the class of drugs which function by deterring some of enzymes of the phosphoinositide 3-kinase, which not only constitute the major part of the pathway but also are an important signaling component for many cellular functions. PTEN (phosphatase and tensin homologue that is deleted in chromosome 10), 4E-binding protein 1 (4EBP1), TSC (tuberous sclerosis complexes 1/2), the downstream substrate of the pathway S6 kinase 1 have been shown to have the capability of phosphorylating the activation function domain 1 of the ER.
Importance of P13K/AKT/mTOR signaling pathway with respect to breast cancer
The process of PI3K/AKT/mTOR pathway activation is initiated by the enzymatic receptor stimulation, which further influences the process of cell proliferation, differentiation, apoptosis, and angiogenesis as well. Gene mutations in PI3K pathway during breast cancer are known to play an important role that encode catalytic and regulatory subunits in the pathway.70 The mutations in HER 2 (RTKs receptors) upon phosphorylation activate the PI3K/AKT/mTOR pathway, while the AKT activation may result in hormonal therapy resistance. There are some of the key mTOR inhibitors enlisted in Table 1, which result in the inhibition of the tumor growth. These mTOR inhibitors play an indispensable role in malignant cell inhibition. Some of the studies demonstrate that the inhibitors to mTOR pathway in combination with other drugs could significantly reduce the side effects along with superior efficacy of these inhibitory molecules.71 The proposed mechanism of action of inhibitors has been elucidated in Fig. 3. These mTOR inhibitors are reported to block the binding of the accessory protein, raptor to mTOR, for example, Metformin and resveratrol that act through the upstream pathway upon the mTOR. Similarly, rapamycin and rapalogs displayed direct inhibition of mTOR. Moreover, Sirolimus, an another inhibitor, also suppresses the renal, hepatic, and cardiac tissue cellular respirations.72
Conclusions and Future Perspectives
Present review has done thorough investigations into various preclinical and clinical studies, where a number of inhibitors of PI3K/Akt/mTOR pathway have been used either alone or in combination with other agents for the treatment of cancer. Basic and targeted agents that are currently under development have been discussed altogether for the better cure of cancer. Efforts have been made to understand the structure–function relationship and regulation/deregulation of mTOR, along with modern generation inhibitors for PI3K/AKT/mTOR pathway besides targeted therapies. mTOR inhibitor studies have clearly revealed their role in antitumor activities as a result of either the activation of different components or some alterations in the gene expression levels. Clearly, the role of mTOR inhibitory drugs in the breast cancer treatment needs to be understood more precisely for specific targeting. Simultaneously, there is a strong need to understand the signaling mechanism in tumor cell along with its behavior, which will certainly add value to assist in managing the diverse treatment methods. Moreover, it will help building in a precise and accurate platform for the personalized medicines paving a way for understanding dreadful diseases like cancer for their proper management. mTOR inhibitor development needs to be further encouraged, which might prove useful to control malignancies in future.
Footnotes
Acknowledgments
We sincerely apologize to those whose work could not be cited owing to space limitations. The authors are grateful to the M.M. (Deemed to be University) for providing the requisite platform to write this article.