
Abstract
Background:
Photobiomodulation therapy (PBMT) using devices to deliver red and/or near-infrared light to tissues has shown promising effects in clinical settings for respiratory diseases, including potential benefits in managing symptoms associated with COVID-19.
Objective:
To determine if at-home self-administered PBMT for patients with COVID-19 is safe and effective.
Methods:
This was a randomized controlled trial (RCT) carried out at home during the COVID-19 pandemic (September 2020 to August 2021). The treatment group self-administered the Vielight RX Plus PBMT device (635 nm intranasal and 810 nm chest LEDs) and were monitored remotely. Eligible patients scored 4–7 (out of 7) for severity on the Wisconsin Upper Respiratory Symptom Survey (WURSS-44). Patients were randomized equally to Control group receiving standard-of-care (SOC) only or Treatment group receiving SOC plus PBMT. The device was used for 20 min 2X/day for 5 days and, subsequently, once daily for 30 days. The primary end-point was time-to-recovery (days) based on WURSS-44 question 1, “How sick do you feel today?”. Subgroup analysis was performed, and Kaplan–Meier and Cox Proportional Hazards analysis were employed.
Results:
One hundred and ninety-nine eligible patients (18–65 years old) were divided into two subgroups as follows: 136 patients with 0–7 days of symptoms at baseline and 63 patients with 8–12 days of symptoms. Those with 0–7 days of symptoms at baseline recovered significantly faster with PBMT. The median for Treatment group was 18 days [95% confidence interval (CI), 13–20] versus the Control group 21 days (95% CI, 15–28), p = 0.050. The treatment:control hazard ratio was 1.495 (95% CI, 0.996–2.243), p = 0.054. Patients with symptom duration ≥7 days did not show any significant improvement. No deaths or severe adverse events (SAEs) occurred in the Treatment group, whereas there was 1 death and 3 SAEs requiring hospitalization in the Control group.
Conclusions:
Patients with ≤7 days of COVID-19 symptoms recovered significantly faster with PBMT compared to SOC. Beyond 7 days, PBMT showed no superiority over SOC.
Trial Registration:
ClinicalTrials.gov NCT04418505.
Introduction
Although the federal coronavirus disease 2019 (COVID-19) public health emergency has ended in the United States, 1 thousands are still hospitalized each week with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections. As at January 2024, the US Centers for Disease and Control has predicted that the number of daily COVID-19 hospital admissions will remain stable or have an uncertain trend, with 1500 to 9800 daily COVID-19 hospital admissions likely by January 29, 2024. 2 The U.S. National Institutes of Health had reported that when infections do occur, some people have sought “alternative” treatments to treat COVID-19. 3 This is also supported by other reports. 4 Further, the possibility of using a personal home-use device for an infectious disease justifies considering devices for treatment.
The objective of this study was to evaluate whether a home-use photobiomodulation (PBM) device could accelerate the time-to-recovery from acute moderate-to-severe SARS-CoV-2 infection. The device is complaint with Health Canada requirements for safety and efficacy. 5
Red and near infrared (NIR) light is delivered to the upper respiratory tract (through the nasal cavity) and the lower respiratory tract (through the sternum). This irradiates the affected parts of the body because the infection starts in the upper respiratory tract and quickly progresses to the lower respiratory tract. PBM modulates the activities of the mitochondrial electron transport chain (ETC). 6 This mechanism may have a role in the treatment of acute COVID-19 disease by systemically improving the immune system function. 7 In addition, PBM can dissociate nitric oxide (NO) from cytochrome c oxidase in the ETC of the mitochondria. 8 NO has been shown to inhibit the replication of exposed coronavirus variants, of which SARS-CoV-2 is one example. 9 –13 Moreover, NO production is also increased by the phosphorylation of endothelial nitric oxide synthase caused by NIR light. 14 The intranasal delivery of the red light is intended to inhibit the virus in the upper respiratory tract, whereas the NIR light delivered to the sternum is meant to inhibit the virus and aid with recovery of the lower respiratory tract, primarily the lungs.
The nasal cavity was selected to place a Light Emitting Diode (LED) because of its proximity to the dense networks of blood capillaries, which are covered by a thin, light-transmitting membrane in the nasal mucosa. This allows the light from the RX-Plus intranasal LED to increase blood flow 15 and easily penetrate and reach both the cell-free mitochondria in the bloodstream 16 and those in tissue cells. This exposure is believed to potentially increase the number of T-lymphocytes. 17 Further, since NO can inhibit the replication of coronaviruses, 5 the use of intranasal PBM to release NO could help to prevent the SARS-CoV-2 virus from spreading to the lower respiratory system.
We also targeted the bone marrow in the sternum for PBM irradiation to enhance the release of stem cells. 18 The stem cells could migrate to the thymus gland where they would mature into T-cells. 19 Moreover, PBM could stimulate the thymus gland thus countering the adverse effects of age-related thymic involution. 20 Positioning the LED on the chest area, including the lungs, irradiates sites with many endothelial cells that are damaged in more advanced stages of COVID-19 disease. 21 Catching the progression of the infection at an early stage may mitigate the risk of morbidity and mortality caused by severe endothelial damage. 22 The ability of PBM to support endothelial function 23,24 could prevent or alleviate acute respiratory distress syndrome (ARDS) and impaired pulmonary function, 25 –27 which are features of acute COVID-19 disease. 7,28,29 In addition, NIR light may improve other biological functions in the thoracic areas by virtue of its ability to accelerate tissue healing, 30 as well as boosting immune system activity through the release of macrophages. 31
Severe SARS-CoV-2 infections are associated with dysregulation of inflammatory immune responses, which in turn inhibits the development of host protective immunity to the infection. Therefore, the use of approaches that modulate inflammation without compromising the adaptive immune response could be an effective therapeutic strategy. 32 For these reasons, PBM may be an effective intervention in attenuating the inflammation caused by COVID-19 disease. This approach has been investigated in animal studies, 33 –35 as well as in human studies, by delivering NIR light to the chest area. 36 In addition, PBM has been shown to reduce the elevated cell count in bronchoalveolar lavage, inflammatory cytokines, and neutrophil numbers. 37
Various case reports, 37 –39 as well as pilot studies, 18,31,40 have suggested the ability of PBM to treat acute COVID-19 disease, as supported by several reviews in the published literature. 15,41 –45 The published literature taken as a whole suggests that PBM using the correct set of parameters may be able to treat acute COVID-19 and, therefore, warranted undertaking this RCT.
Materials and Methods
Trial design
This was a parallel, multi-center, randomized clinical trial designed to test the effectiveness of a home-use PBM device to provide COVID-19 symptom relief measured by decreased time of symptoms (days). The Control group received government-recommended standard of care (Control), whereas the Treatment group additionally received self-administered home treatment with the stand-alone PBM device (Treatment). The trial was designed to gather data of effectiveness in a home-use environment. The trial complied with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. The protocol was approved by Health Canada and an institutional review board. Patients provided signed informed consent before enrollment. Information was posted on ClinicalTrials.gov NCT04418505 before the start of the trial.
COVID-19 symptom improvement was measured based on the response to relevant questions (Q1-Q43) on the Wisconsin Upper Respiratory Symptom Survey (WURSS)-44, scoring from 0 (not sick) to maximum 7 (severely sick) (Supplementary Data S1).
Each patient uploaded their answers daily using the REDCap Cloud electronic data capture (EDC) platform for 30 days.
During the RCT, an intervention device was shipped to each treatment patient within 24 hours of randomization.
Patients and procedures
Patients were recruited according to the following inclusion and exclusion criteria:
Inclusion criteria
Confirmation of SARS-CoV-2 with reverse transcriptase–polymerase chain reaction (PCR) test or alternative WURSS-44 Question 1 score of 4–7 Between 18 and 65 years of age
Exclusion criteria
Need for hospitalization at the time of diagnosis Current need for supplemental oxygen or positive pressure support and/or has required supplemental oxygen or positive pressure support for > or = 24 hours >10 days from symptom onset Pregnant Inability to complete daily study forms online (in English) Diagnosis of Chronic Obstructive Pulmonary Disease Positive for Hepatitis C Virus, Hepatitis B Virus, or Human Immunodeficiency Virus Inability to speak English
Note that the inclusion score of 4–7 on WURSS Q1 on Day 1 of treatment was used as the baseline for efficacy analysis. Study participants included those with a baseline score of ≥4 for WURSS Q1, indicative of moderate-to-severe severity, to allow a measurable improvement from baseline. Given that the study was not blinded, we intentionally excluded patients with mild symptoms (<4) who may be more susceptible to placebo effects and other unforeseen factors.
Patients were registered using EDC software and then randomized with equal allocation to the Treatment or Control group using the OxMAR minimization software (Fig. 1). 46 The first patient was recruited on September 20, 2020. The 294th patient who was also the last was recruited on July 5, 2021. Each patient received compensation of US$500.
A Consolidated Standards of Reporting Trials (CONSORT) diagram presenting each stage of the randomized trial is shown in Fig. 1.
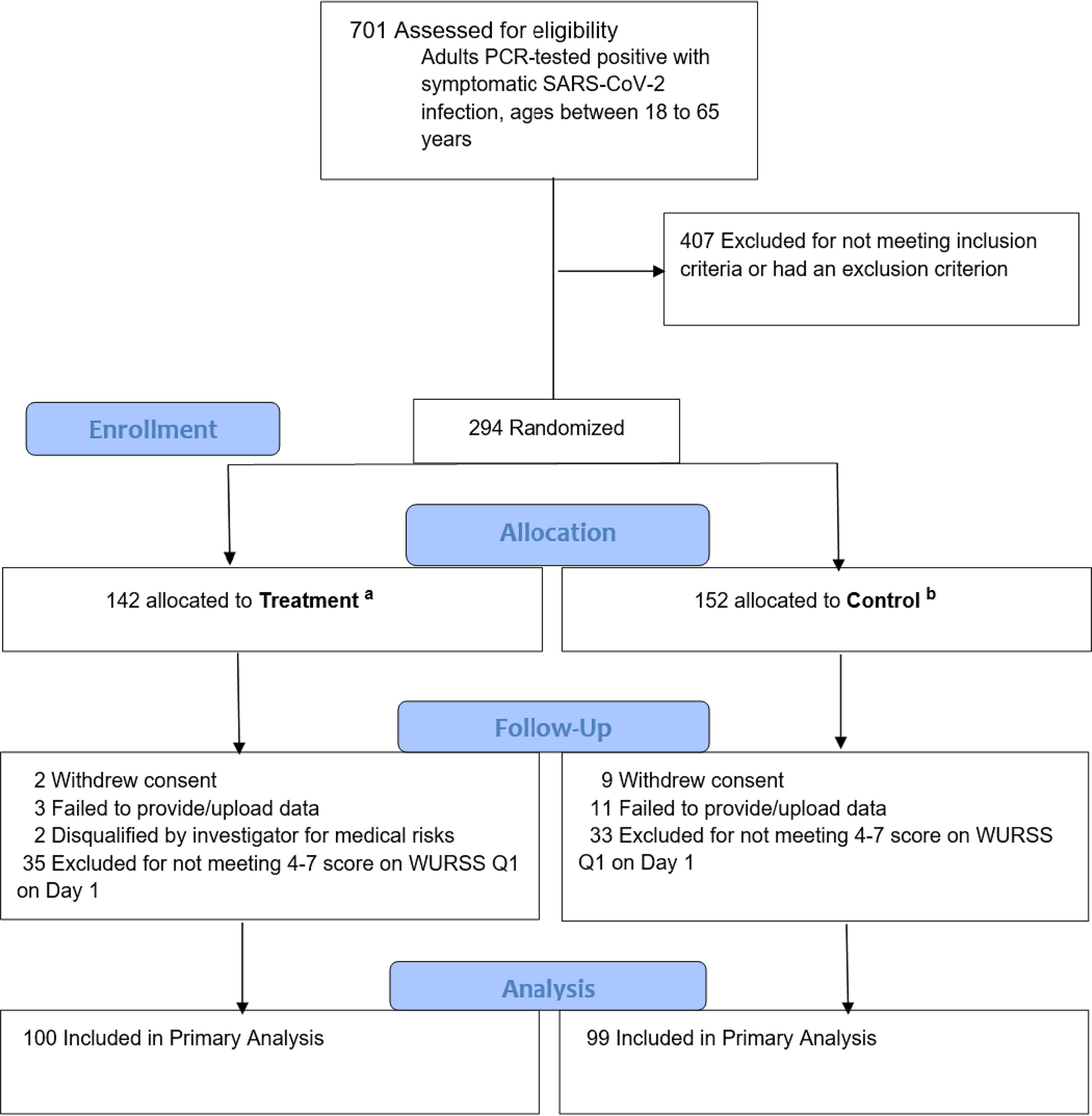
CONSORT diagram. Flowchart of participants through each stage of the randomized trial.
Trial intervention device and monitoring
The PBM intervention device called the “Vielight RX Plus” is shown in Figure 2. It consists of a near infrared (NIR) light emitting diode (LED) module that is positioned on the sternum and thymus gland (“Sternum & thymus LED module”), a red LED intranasal applicator, and a controller. The LED module and applicator are controlled by a driver, which is connected to a power source. The device is portable and suitable for use at home.
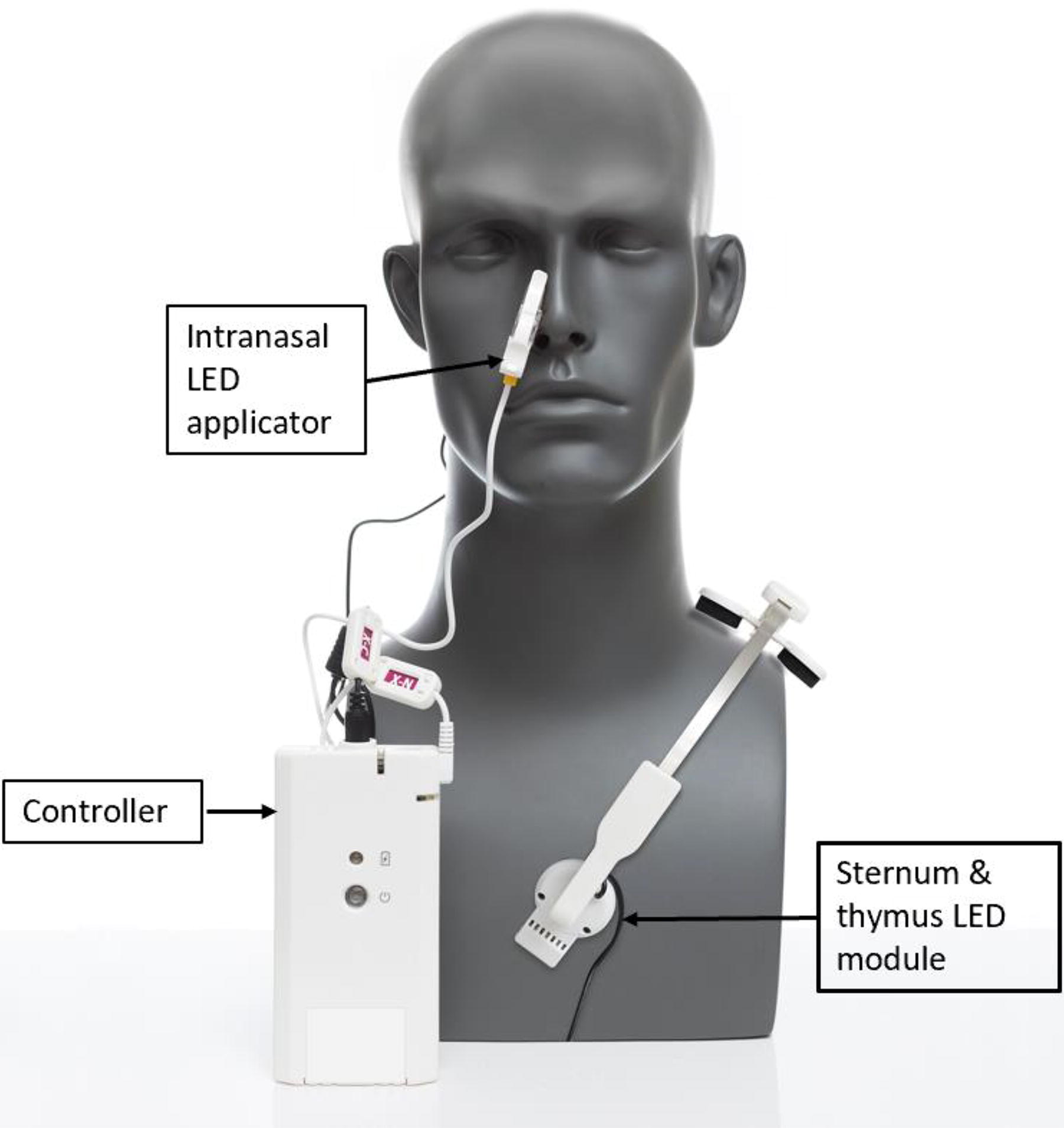
The portable and home-use Vielight RX Plus intervention device. The specifications for the device are set out in Table 1.
The specifications of the intervention device are set out in Table 1.
Vielight RX Plus Specifications
The treatment was self-administered for 20 min twice a day for the first 5 days and, subsequently, once daily for the remaining 25 days. A pulse oximeter was shipped to each patient to measure oxygen saturation.
Efficacy end-points
The primary efficacy end-point was the time-to-recovery (days) as determined by WURSS-44 Q1, “How sick do you feel today?”. Successful recovery was defined as the first day of 3 consecutive days with a 0 (not sick) score.
Secondary efficacy end-points included the time-to-recovery (days) for WURSS-44 Q2–Q43 and the number of days with mild symptoms (0–3 WURSS-44 scores). The questions include the following: 2) cough, 3) coughing up stuff, 4) cough interfering with sleep, 5) sore throat, 6) scratchy throat, 7) hoarseness, 8) runny nose, 9) plugged nose, 10) sneezing, 11) headache, 12) body aches, 13) feeling ‘run down’, 14) sweats, 15) chills, 16) feeling feverish, 17) feeling dizzy, 18) feeling tired, 19) irritability, 20) sinus pain, 21) sinus pressure, 22) sinus drainage, 23) swollen glands, 24) plugged ears, 25) discomfort, 26) watery eyes, 27) eye discomfort, 28) head congestion, 29) chest congestion, 30) chest tightness, 31) heaviness in chest, 32) lack of energy, 33) lack of appetite, 34) difficulty in thinking clearly, 35) difficulty in speaking clearly, 36) poor sleeping, 37) difficult breathing, 38) walk, climb stairs, exercise, 39) accomplish daily activities, 40) work outside the home, 41) work inside the home, 42) interact with others, and 43) live your personal life.
We excluded Q44, “Compared to yesterday, I feel that my cold is …” because it was not specific to COVID-19.
The full WURSS-44 Daily Symptom Report can be viewed in Supplementary Data S1.
Safety assessments included evaluating the frequency and percentage of patients experiencing adverse events (AEs). To further ensure safety, patients were equipped with pulse oximeters to monitor their oxygen saturation levels, ensuring that they remained above 90% and report SpO2 data daily.
Sample size calculation
The trial aimed for a sample size of 280 patients, with a 1:1 randomization. This was calculated to detect a hazard ratio (HR) between 1.40 and 1.44, ensuring 80% statistical power at a 5% significance level. This HR assumption was based on the expected treatment effect and the anticipated event rate in the control group. The calculation accounted for the necessary detectable effect size with adequate power, considering potential dropouts. SAS software was used to validate these parameters, ensuring that our study was appropriately powered to assess the treatment efficacy.
Statistical methods
Time-to-recovery (days) was estimated with the Kaplan–Meier (KM) method overall and separately for 0–5 days and 6–10 days of symptom duration strata. 47 The symptom duration was the number of days each patient had COVID-19 symptoms by the time of enrollment. A log-rank test stratified by duration of symptoms (0–5 days, >5 days) compared the efficacy end-point median distributions between Treatment and Control by symptom duration. An unstratified KM method and log-rank test were used to evaluate median time-to-recovery within the symptom duration strata. Subjects who did not recover within 30 days were censored on Day 30. Supportive analyses included stratified and unstratified Cox Proportional Hazards models with 95% confidence intervals (CIs). 48 Subgrouping by symptom duration strata was a post hoc analysis.
An analysis of variance (ANOVA) was used to compare mean days of mild symptoms with terms for treatment and symptom duration strata. Unstratified analyses were by ANOVA with Treatment as the explanatory variable. A linear mixed model repeated measures analysis of covariance was used to compare percentage changes in oxygen saturation in safety monitoring. 49 Model terms included treatment, days (7, 14, 21, 28), symptom days strata treatment-by-day interaction, and baseline covariate.
Frequency distributions of adverse events (AEs) were presented. A Poisson regression model was used to compare the mean number of episodes of AE and the mean number of patients with AEs, between Treatment and Control. 50
An interim analysis was conducted in January 2021. The results from 73 patients indicated that the study should continue and not be stopped for futility nor superiority (Supplementary Data S1).
We did not impute missing data. Instead, we used Kaplan–Meier estimates, which accommodate varying follow-up durations, assuming that any censoring is noninformative.
SAS software was used for statistical analysis. 16 All p-values were two-sided, and a p value < = 0.050 was used to signify statistical significance.
Results
Patients
Recruitment started in September 2020. A total of 701 adults who tested positive for COVID-19 were assessed for eligibility. Of these, 407 failed the initial inclusion/exclusion criteria. 406 participants failed to meet the inclusion criterion of severity scores of between 4 and 7 for Question 1 of the WURSS-44 scale, “How sick do you feel today?” which would have indicated a moderate-to-severe level of sickness; or the participants were younger or older than the qualifying ages of between 18 and 65. One candidate was unable to fill the forms in English, which was an exclusion criterion. This left 294 patients eligible for enrollment, randomization, and allocation. Due to factors such as the increased availability of monoclonal antibodies for treatment, declining interest in the study, and having achieved sufficient numbers for statistical power, we decided to cease recruitment on July 5, 2021 and locked the datasets of the 294 qualified participants.
Subsequently, 27 patients were lost during follow-up study qualification, leaving 267 for AE assessment.
Shipping added a mean of 2 days, extending the 0–5 symptom days stratum to 0–7 days and 6–10 symptom days stratum to 8–12 days. Therefore, the strata of symptom duration at baseline for efficacy analyses should be interpreted as 0–7 days and 8–12 days.
During the 2-day shipping period, 35 in Treatment and 33 in Control were excluded from efficacy analysis for scoring below the qualifying score of 4 on the WURSS-44 severity scores due to spontaneous improvement. The remaining 199 patients in the intention-to-treat population consisted of 100 Treatment and 99 Control patients. See Figure 1.
Demographics, baseline characteristics, and WURSS-44 severity scores of the 199 patients in efficacy analysis are presented in Table 2.
Patient Demographics, Baseline Characteristics, and Symptom Severity Scores
BMI, body mass index; IQR, interquartile range; WURSS, Wisconsin Upper Respiratory Symptom Survey.
Race and ethnicity were self-reported and not considered for deeper analysis.
Calculated as weight in kilograms divided by height in meters squared.
Patients were stratified by symptom duration strata 0–7 days or 8–12 days of having COVID-19 symptoms on Day 1 of Treatment (Baseline of efficacy analyses).
Qualifying patients at Baseline had WURSS-44 Q1 scores of 4–7.
Primary efficacy outcomes
Among all subjects (n = 199), median days to recovery was 19 days for active and 21 days for control, which were not statistically significant (difference +2 days, p = 0.197 log-rank, HR 1.25 p = 0.199 Cox).
For the 0–7 symptom days stratum (n = 136), median days to recovery was 18 days for active and 21 days for control, which approached statistical significance (difference +3 days, p = 0.050 log-rank, HR 1.495 p = 0.052 Cox).
For the 8–12 symptom days stratum (n = 63), median days to recovery was in the opposite direction with 23 days for active and 21 days for control (difference −2 days, p = 0.507 log-rank, HR 0.803 p = 0.499 Cox).
The overall HR of 1.495 was consistent with the planned target HR. The KM curve of the probability of not recovering over the 30-day assessment period for the 0–7 symptom days stratum is presented in Figure 3.
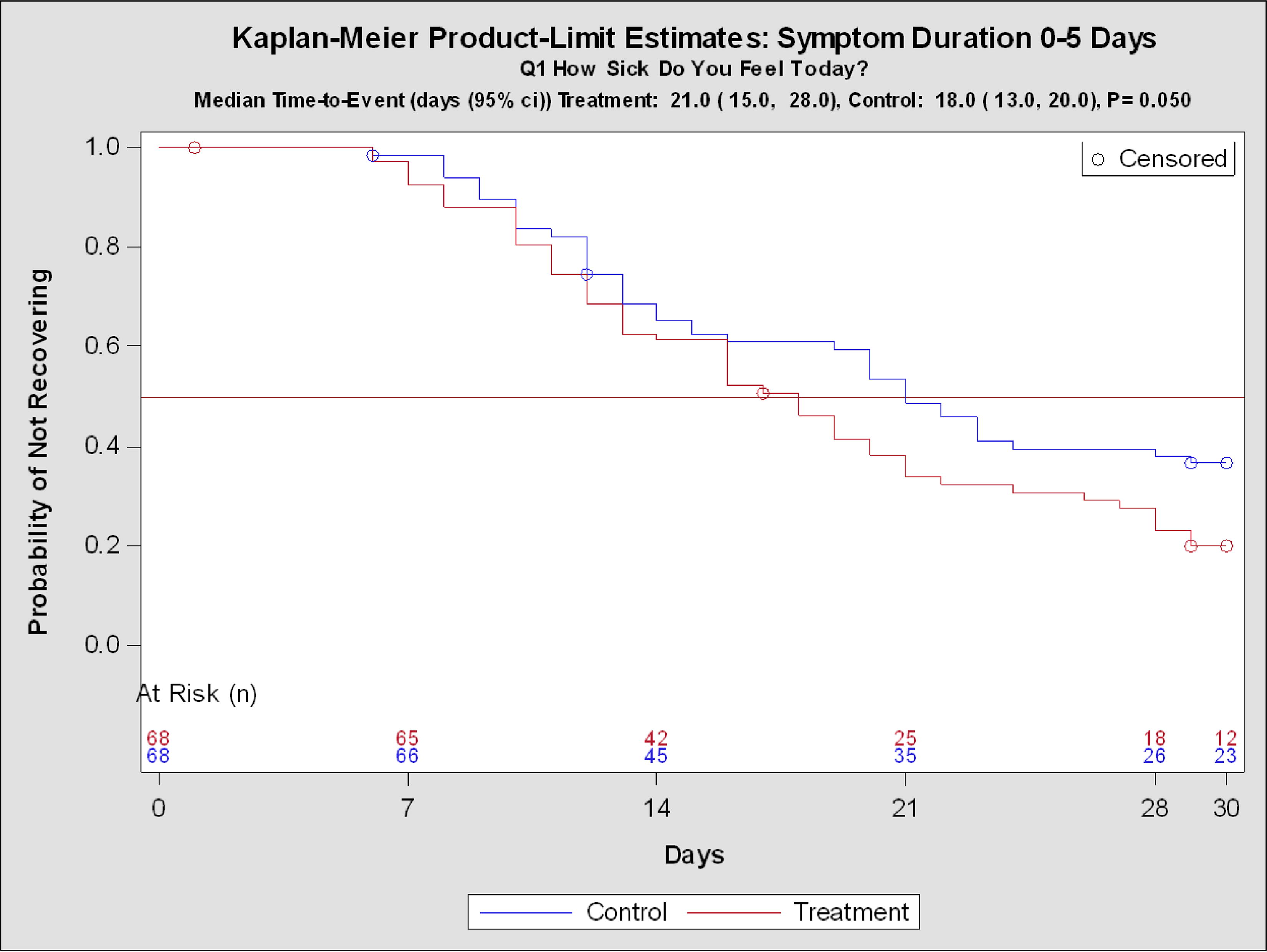
Kaplan–Meier Curve of Primary Outcome. Symptoms duration of 0–7 days at Baseline. “How Sick Do You Feel Today?” (WURSS-44 Q1). This figure shows the probability of not recovering from the WURSS-44 Q1 symptom of “How sick do you feel today?”. It represents the outcome for the stratified group with symptom duration of 0–7 days at Baseline (0–5 days on Enrollment). Subjects that did not have full recovery were censored.
Secondary efficacy outcomes
As secondary efficacy outcomes, patients with WURSS-44 Q1 severity scores of 4–7 at Baseline were assessed for time-to-recovery for Q2–Q43 (Table 3). For the full population, statistical significance favoring Treatment in the time-to-recovery was observed for sinus pain, chest congestion, body aches, thinking clearly, ear discomfort, sinus drainage, headache, coughing up sputum, and sneezing. Control was not superior to Treatment in any of the WURSS-44 questions.
Secondary Outcomes of Patients with p < 0.050 by Symptom Duration Strata
The symptom duration represents the number of days that the patients experienced symptoms when treatment started on Day 1 (Baseline).
Kaplan–Meier (KM) method curves are presented in Supplement 2, eFigure 2.
For the 0–7 symptom days duration stratum, significance was observed for headache, sinus pain, thinking clearly, swollen glands, chest congestion, sinus drainage, and body aches. Control was not superior to Treatment in any of the WURSS-44 questions.
For the 8–12 symptom days stratum, the Treatment group recovered significantly more slowly from feeling tired, runny nose, and lack of energy.
We also assessed the Treatment effectiveness in reducing symptom severity, expressed as the mean number of days of mild symptoms (WURSS-44 Q1–43 scores of 0–3). For all patients, Treatment results were significantly better for runny nose, sneezing, body aches, irritability, and ear discomfort. For the 0–7 days symptom duration stratum, the Treatment group showed significant improvement in headache. For the 8–12 days stratum, the Treatment group showed significant improvement for runny nose, sneezing, body aches, sinus drainage, and plugged ears. Control was not superior to Treatment in any WURSS-44 questions overall or by symptom duration strata.
Control was superior to Treatment in time-to-recovery for tiredness, energy level, and runny nose in the 8–12 days symptom duration stratum. Runny nose is an expected minor side effect arising from intranasal PBM relieving sinus congestion through improving blood circulation in the sinuses.
Adverse events
After initial follow-up, 267 enrolled patients (135 in Treatment and 132 in Control) were monitored for adverse events. The population eligible for safety monitoring included all randomized subjects with a response of ≥4 in severity score for WURSS Q1 on enrollment.
None of the Treatment patients suffered death or severe adverse events (SAEs). In Control, there were 4 (3.0%) SAEs that required hospitalization, including 1 death. These hospitalized patients exhibited respiratory symptoms, and the one recorded death was attributed to a range of symptoms associated with COVID-19.
AEs occurring in >5% of patients are listed in Table 4. In comparison with the Control group, patients in Treatment had significantly fewer patients with the AEs of tachycardia and dysgeusia (altered taste) but not for other AEs. For tachycardia, it was 14.30% (95% CI, −25.9%- −1.87%), p = 0.016. For dysgeusia (altered taste) it was 13.00% (95% CI, −21.20-−4.63%), p = 0.001.
Adverse Events Not Requiring Hospitalization
Only symptoms observed in over 5% of monitored patients are listed. A total of 267 patients were monitored for adverse events post enrollment.
In the assessment of oxygen saturation changes, Treatment did not show any worsening in comparison with the control group (97.69% for Treatment vs. 97.36% for Control).
Discussion
The primary end-point of recovery (WURSS-44 Q1) from general sickness in COVID-19 infection after treatment was dependent on the duration of the symptoms in those with moderate-to-severe symptoms at baseline. Based on the two separate strata of 0–7 days and 8–12 days of symptom duration, Treatment patients who presented 0–7 days of symptoms experienced significantly faster recovery than patients on SOC alone (Control) (p = 0.050). Patients with longer symptom duration of 8–12 days did not show a statistically significant difference in the primary efficacy end-point outcome.
This study was conducted from September 2020 to July 2021, during which the U.S. and Canadian government guidelines on SOC for treating nonhospitalized SARS-CoV-2 patients varied. These SOC guidelines included home isolation, mask-wearing, vaccination, and, where available, the use of monoclonal antibodies. The changing nature of SOC should not significantly influence the study results because both the Treatment group (who received PBM therapy in addition to SOC) and the Control group had equal access to the recommended SOC.
Based on the primary end-point, the Vielight RX Plus can be indicated for achieving significantly faster recovery for patients with acute COVID-19 infection and up to 7 days of symptoms of moderate-to-severe levels of severity.
PBM did not produce positive results in the 8–12 symptom day stratum as shown by statistically nonsignificant results in this stratum. The lack of response could be due to the observation that in acute and severe cases of SARS-CoV-2 infection, approximately 20% of patients experience a breakdown of the epithelial barrier. This breakdown can result in significant endothelial damage and a swift decline in health, potentially necessitating mechanical ventilation. 17 This extensive damage may render PBM ineffective when delivered with the present device specifications of low power.
In general, patients in the Treatment group improved significantly better than the Control group in most respiratory symptoms. Treated patients with 0–7 days of symptom duration experienced quicker recovery than Control for headache, sinus pain, thinking clearly, swollen glands, chest congestion, and body aches and experienced more days with milder headaches. Treated patients with 8–12 days of symptoms experienced slower recovery from tiredness, runny nose, and energy deficits but experienced more days with milder levels of severity for runny nose, sneezing, body aches, sinus drainage, and plugged ears.
Significantly fewer treated patients experienced tachycardia and dysgeusia (altered taste), which were the most frequent severe adverse events reported. The oxygen saturation levels did not show worsening in the Treatment group compared with the Control group.
It is likely that many mechanisms of PBM played a role in the positive outcomes reported in this study. Not least among them were the effect on inflammation, which has been cited as a major cause of hospitalizations. 51 Several clinical PBM studies have focused on inflammation and the related inflammatory cytokines and have shown that PBM resulted in significant reduction in cytokine markers. 34,37 One clinical study on 50 COVID-19 patients used flexible pads with 650 nm plus 850 nm LEDs to deliver light to 1000 cm2 of the body surface, concentrating on the chest and sinuses. 31 One to three 84-minute exposures produced improvements in many COVID symptoms, especially if the time between the onset of symptoms and PBMT was less than 5 days. Another clinical study with 10 patients (5 treatment and 5 control) used an MLS scanner-equipped laser (808 nm and 905 nm) to deliver light to the chest daily for four days. 18 The treatment group recovered faster than the control group and did not require admission to the ICU, whereas in the control group 3 patients were admitted to ICU and two of them died.
Various studies reporting the effect of released NO on coronaviruses within a genotype family suggest that PBM efficacy in treating coronavirus disease is not variant specific. 13,24 –26 For this reason, PBM may be considered as a potential remedy for future variants of SARS-CoV-2. However, this hypothesis requires further testing.
This RCT had several limitations. First, it was not double-blinded, and a placebo or sham device was not used due to the visible red light of the device, which made blinding ineffective. Second, our methodology relied on self-reporting with the WURSS-44 questionnaire, which was necessary for safe data collection during the pandemic. Third, the sample size available for each secondary efficacy end-point (WURSS-44 Q2–43) was as expected much smaller compared to the aggregate number for the primary end-point of general sickness. Consequently, the statistical power for these secondary end-points would be inherently lower.
This RCT used a low-powered, convenient home-use device. Higher powered pulsed lasers may produce improvements in very long-standing severe patients. The lower power density of the home-use LED device used in this study may not have been sufficient to effectively treat severe infections with symptom durations exceeding 7 days. This opens an avenue for future research to investigate higher power densities and other parameters to enhance the effectiveness of PBM devices in similar studies for COVID-19.
Today, long COVID is still a major public health issue. 52 A recent study suggests that PBM could be promising for treating brain fog in long COVID. 53 This RCT also suggests that PBM devices of appropriate design and delivering the right parameters could be effective in treating long COVID. A study for long COVID has been planned to follow this work, being motivated by the improvement in thought clarity (WURSS-44 question on “thinking clearly”) to primarily target improvements in brain fog. 54
Conclusions
In conclusion, COVID-19 patients experiencing symptoms for up to 7 days achieved a significantly quicker overall recovery and experienced fewer days of milder headaches when treated with PBM using the Vielight RX Plus home-use device, in addition to standard of care (SOC), compared with those receiving SOC alone. They also experienced quicker recovery from headache, sinus pain, deficit in thinking clearly, swollen glands, chest congestion, and body aches. Patients with more than 7 days of symptoms were shown to recover more slowly with the PBM treatment, although not significantly so. However, they could expect more days of milder symptoms for runny nose, sneezing, body aches, sinus drainage, and plugged ears.
The results of this RCT suggest that PBM also has a potential role in treating long COVID, a pressing and unresolved public health issue. Ideally, a double-blind placebo-controlled randomized trial should be carried out, but nevertheless these findings provide a basis for follow-up studies.
Data Availability Statement
The original data presented in the study are included in the Supplementary Data S1; further inquiries can be directed to the corresponding author.
Ethics Statement
The study which involved human participants was reviewed and given Investigational Testing Authorization by the Medical Devices Directorate of Health Canada (application No. 315768) and reviewed and approved by an independent institutional review board, Advarra, Inc. IRB Services, Aurora, ON, Canada. The patients/participants provided their written informed consent to participate in this study.
Footnotes
Acknowledgments
Vielight Inc. contributed all the Vielight RX Plus devices and the oximeters used in the study. The authors thank Bruce Barrett, MD, PhD of the University of Wisconsin—Madison, for his valuable insight and use of the WURSS-44 survey instrument in the clinical trial. The authors thank Conrad Kabali, PhD, for additional third-party statistical analysis and advice, Hilary Au, PhD, for the management of the shipping and logistics, and other staff of Vielight for follow-through support involved in the clinical trial.
Author Contributions
L.L. conceived the initial study based on a white article by L.L. and M.R.H. K.F.H. and M.V.B. developed the statistical analysis plan. L.L., N.H., K.F.H., G.L., Z.A., M.K., A.S., and A.P. designed the study and developed the protocol. A.B., N.H., C.H., M.Z., A.W., J.R., B.Z., R.G.C., D.R.T., and D.J.M. were responsible for study enrollment and data acquisition. L.L. was responsible for financial management. N.H. and A.B. were responsible for operations and database management. M.V.B., K.F.H., L.L., and N.H. were responsible for statistical analyses and the interpretation of the data. A.B., C.H., and N.H. verified the underlying data. LL drafted the article and provided the scientific bases. M.V.B., N.H., and M.R.H. critically reviewed the article. L.L., N.H., and A.B. have full access to the data and their accuracy and integrity. All authors contributed to conducting the trial, revised the report, and read and approved the final version before submission. LL obtained the funding.
Author Disclosure Statement
L.L. was shareholder and chief executive officer of Vielight Inc.; N.H., G.L., Z.A., M.K., A.S., and M.K. were employees of Vielight Inc. at the time of the clinical trial. Data Reduction LLC, Impact Clinical Trials Marketing & Management Services, Ironstone Product Development, Progressive Medical Research, MKR Clinical Research Consultants, Malton Medical Clinic, Aeremedea LLC, Tidal Rehabilitation Center, and Pennsbury Medical Practice received fees from Vielight Inc. for various services rendered during the clinical trial. M.V.B., A.B., K.F.H., A.P., M.Z., A.W., J.R., B.Z., C.H., R.G.C., D.R.T., and D.J.M. were principals, partners, or employees of these organizations. M.R.H. received advisory fees from Vielight Inc.
Funding Information
This study was entirely funded by Vielight Inc.
Supplementary Material
Supplementary Data S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.